Folding funnel
The folding funnel hypothesis is a specific version of the energy landscape theory of protein folding, which assumes that a protein's native state corresponds to its free energy minimum under the solution conditions usually encountered in cells. Although energy landscapes may be "rough", with many non-native local minima in which partially folded proteins can become trapped, the folding funnel hypothesis assumes that the native state is a deep free energy minimum with steep walls, corresponding to a single well-defined tertiary structure. The term was introduced by Ken A. Dill in a 1987 article discussing the stabilities of globular proteins.[1]
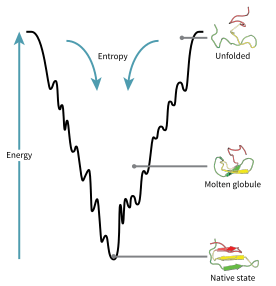
The folding funnel hypothesis is closely related to the hydrophobic collapse hypothesis, under which the driving force for protein folding is the stabilization associated with the sequestration of hydrophobic amino acid side chains in the interior of the folded protein. This allows the water solvent to maximize its entropy, lowering the total free energy. On the side of the protein, free energy is further lowered by favorable energetic contacts: isolation of electrostatically charged side chains on the solvent-accessible protein surface and neutralization of salt bridges within the protein's core. The molten globule state predicted by the folding funnel theory as an ensemble of folding intermediates thus corresponds to a protein in which hydrophobic collapse has occurred but many native contacts, or close residue-residue interactions represented in the native state, have yet to form.
In the canonical depiction of the folding funnel, the depth of the well represents the energetic stabilization of the native state versus the denatured state, and the width of the well represents the conformational entropy of the system. The surface outside the well is shown as relatively flat to represent the heterogeneity of the random coil state. The theory's name derives from an analogy between the shape of the well and a physical funnel, in which dispersed liquid is concentrated into a single narrow area.
Background
The protein folding problem is concerned with three questions, as stated by Ken A. Dill and Justin L. MacCallum: (i) How can an amino acid sequence determine the 3D native structure of a protein? (ii) How can a protein fold so quickly despite a vast number of possible conformations (the Levinthal's Paradox)? How does the protein know what conformations not to search? And (iii) is it possible to create a computer algorithm to predict a protein's native structure based on its amino acid sequence alone?[2] Auxiliary factors inside the living cell such as folding catalysts and chaperones assist in the folding process but do not determine the native structure of a protein.[3] Studies during the 1980s focused on models that could explain the shape of the energy landscape, a mathematical function that describes the free energy of a protein as a function of the microscopic degrees of freedom.[4]
After introducing the term in 1987, Ken A. Dill surveyed the polymer theory in protein folding, in which it addresses two puzzles, the first one being the Blind Watchmaker's Paradox in which biological proteins could not originate from random sequences, and the second one being Levinthal's Paradox that protein folding cannot happen randomly.[5] Dill pulled the idea from the Blind Watchmaker into his metaphor for protein folding kinetics. The native state of protein can be achieved through a folding process involving some small bias and random choices to speed up the search time. That would mean even residues at very different positions in the amino acid sequence will be able to come into contact with each other. Yet, a bias during the folding process can change the folding time by tens to hundreds of orders of magnitude.[5]
As protein folding process goes through a stochastic search of conformations before reaching its final destination,[3] the vast number of possible conformations is considered irrelevant, while the kinetic traps begin to play a role.[5] The stochastic idea of protein intermediate conformations reveals the concept of an “energy landscape” or "folding funnel" in which folding properties are related to free energy and that the accessible conformations of a protein are reduced as it approaches native-like structure.[3] The y-axis of the funnel represents the "internal free energy" of a protein: the sum of hydrogen bonds, ion-pairs, torsion angle energies, hydrophobic and salvation free energies. The many x-axes represent the conformational structures, and those that are geometrically similar to each other are close to one another in the energy landscape.[6] The folding funnel theory is also supported by Peter G Wolynes, Zaida Luthey-Schulten and Jose Onuchic, that folding kinetics should be considered as progressive organization of partially folded structures into an ensemble (a funnel), rather than a serial linear pathway of intermediates.[7]
Native states of proteins are shown to be thermodynamically stable structures that exist in physiological conditions,[3] and are proven in experiments with ribonuclease by Christian B. Anfinsen (see Anfinsen's dogma). It's suggested that because the landscape is encoded by the amino-acid sequence, natural selection has enabled proteins to evolve so that they are able to fold rapidly and efficiently.[8] In a native low-energy structure, there's no competition among conflicting energy contributions, leading to a minimal frustration. This notion of frustration is further measured quantitatively in spin glasses, in which the folding transition temperature Tf is compared to the glass transition temperature Tg. Tf represents the native interactions in the folded structure and Tg represents the strength of non-native interactions in other configurations. A high Tf/Tg ratio indicates a faster folding rate in a protein and fewer intermediates compared to others. In a system with high frustration, mild difference in thermodynamic condition can lead to different kinetic traps and landscape ruggedness.[9]
Proposed Funnel Models
Funnel-shaped Energy Landscape
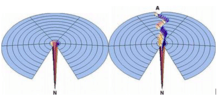
Ken A. Dill and Hue Sun Chan (1997) illustrated a folding pathway design based on Levinthal's Paradox, named the "golf-course" landscape, where a random searching for the native states would prove impossible, due to the hypothetically "flat playing field" since the protein "ball" would take a really long time to find a fall into the native "hole". However, a rugged pathway deviated from the initial smooth golf-course creates a directed tunnel where the denatured protein goes through to reach its native structure, and there can exist valleys (intermediate states) or hills (transition states) long the pathway to a protein's native state. Yet, this proposed pathway yields a contrast between pathway dependence versus pathway independence, or the Levinthal dichotomy and emphasizes the one-dimensional route of conformation.
Another approach to protein folding eliminates the term "pathway" and replaces with "funnels" where it's concerned with parallel processes, ensembles and multiple dimensions instead of a sequence of structures a protein has to go through. Thus, an ideal funnel constitutes of a smooth multi-dimensional energy landscape where increasing interchain contacts correlate with decreasing degree of freedom and ultimately achievement of native state.[6]
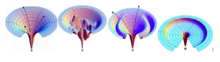
Unlike an idealized smooth funnel, a rugged funnel demonstrates kinetic traps, energy barriers, and some narrow throughway paths to native state. This also explains an accumulation of misfolded intermediates where kinetic traps prevent protein intermediates from achieving their final conformation. For those that are stuck in this trap, they would have to break away favorable contacts that do not lead to their native state before reaching their original starting point and find another different search downhill.[6] A Moat landscape, on the other hand, illustrates the idea of a variation of routes including an obligatory kinetic trap route that protein chains take to reach their native state. This energy landscape stems from a study by Christopher Dobson and his colleagues about hen egg white lysozyme, in which half of its population undergo normal fast folding, while the other half first forms α-helices domain quickly then β-sheet one slowly.[6] It's different from the rugged landscape since there are no accidental kinetic traps but purposeful ones required for portions of protein to go through before reaching the final state. Both the rugged landscape and the Moat landscape nonetheless present the same concept in which protein configurations might come across kinetic traps during their folding process. On the other hand, the Champagne Glass landscape involves free energy barriers due to conformational entropy that partly resembles the random golf-course pathway in which a protein chain configuration is lost and has to spend time searching for the path downhill. This situation can be applied to a conformational search of polar residues that will eventually connect two hydrophobic clusters.[6]
The Foldon Volcano-shaped Funnel Model
In another study, Rollins and Dill (2014) introduces the Foldon Funnel Model, a new addition to previous folding funnels, in which secondary structures form sequentially along the folding pathway and are stabilized by tertiary interactions. The model predicts that the free energy landscape has a volcano shape instead of a simple funnel that is mentioned previously, in which the outer landscape is sloped uphill because protein secondary structures are unstable. These secondary structures are then stabilized by tertiary interactions, which, despite their increasingly native-like structures, are also increasing in free energy until the second-to-last to the last step that is downhill in free energy. The highest free energy on the volcano landscape is at the step with structure just before the native state. This prediction of energy landscape is consistent with experiments showing that most protein secondary structures are unstable on their own and with measured protein equilibrium cooperativities. Thus, all earlier steps before reaching the native state are in pre-equilibrium. Despite its model being different from other models before, the Foldon Funnel Model still divides conformational space into the two kinetic states: native versus all others.[10]
Application
Folding funnel theory has both qualitative and quantitative application. Visualization of funnels creates a communicating tool between statistical mechanical properties of proteins and their folding kinetics.[4] It suggests the stability of folding process, which would be hard to destroy by mutation given maintained stability. To be more specific, a mutation can occur that leads to blockage of a routes to native state, but another route can take over provided that it reaches the final structure.[9]
A protein’s stability increases as it approaches its native state through the partially folded configuration. Local structures such as helices and turns happen first followed by global assembly. Despite a process of trial and error, protein folding can be fast because proteins reach its native structure by this divide-and-conquer, local-to-global process.[2] The idea of folding funnel helps rationalize the purpose of chaperones, in which the re-folding process of a protein can be catalyzed by chaperones pulling it apart and bringing it to a high energy landscape and let it fold again in a random fashion of trials and errors.[6] Funneled landscapes suggest that different individual molecules of the same protein sequence may utilize microscopically different routes to reach the same destination. Some paths will be more populated than others.[2]
Funnels distinguish the basics between folding and simple classical chemical reactions analogy. A chemical reaction starts from its reactant A and goes through a change in structure to reach its product B. On the other hand, folding is a transition from disorder to order, not only from structure to structure. Simple one-dimensional reaction pathway does not capture protein folding's reduction in conformational degeneracy.[4] In other words, folding funnels provide a microscopic framework for folding kinetics. Folding kinetics is described by simple mass action models, D-I-N (on-path intermediate I between denatured D and native N) or X-D-N (off-path intermediate X), and is referred to as the macroscopic framework of folding.[4] Sequential Micropath view represents the mass-action model and explains folding kinetics in terms of pathways, transition states, on and off-path intermediates and what one sees in experiments, and is not concerned with the activity of a molecule or the state of a monomer sequence at a specific macroscopic transition state. Its problem is related to Levinthal's Paradox, or the searching problem.[5] In contrast, funnel models aim to explain the kinetics in terms of underlying physical forces, to predict the microstate composition of those macrostates.
Nonetheless, it proves challenging for computer simulations (energy landscape) to reconcile the "macroscopic" view of mass-action models with "microscopic" understanding of the changes in protein conformation during the folding process. Insights from funnels are not sufficient enough to improve computer search methods. A smooth and funnel-shaped landscape on global scale can appear rough on local scale in computer simulations.[2]
See also
- Chaperone – proteins that assist other proteins with folding or unfolding
- Levinthal paradox
- Protein structure prediction
References
- Dill, Ken A. (1987). Oxender, DL; Fox, CF (eds.). "The stabilities of globular proteins". Protein Engineering. New York: Alan R. Liss, Inc.: 187–192.
- Dill KA, MacCallum JL (November 2012). "The protein-folding problem, 50 years on". Science. 338 (6110): 1042–6. Bibcode:2012Sci...338.1042D. doi:10.1126/science.1219021. PMID 23180855.
- Dobson CM (February 2004). "Principles of protein folding, misfolding and aggregation". Seminars in Cell & Developmental Biology. 15 (1): 3–16. doi:10.1016/j.semcdb.2003.12.008. PMID 15036202.
- Dill KA, Ozkan SB, Shell MS, Weikl TR (June 2008). "The protein folding problem". Annual Review of Biophysics. 37 (1): 289–316. doi:10.1146/annurev.biophys.37.092707.153558. PMC 2443096. PMID 18573083.
- Dill KA (June 1999). "Polymer principles and protein folding". Protein Science. 8 (6): 1166–80. doi:10.1110/ps.8.6.1166. PMC 2144345. PMID 10386867.
- Dill KA, Chan HS (January 1997). "From Levinthal to pathways to funnels". Nature Structural Biology. 4 (1): 10–9. doi:10.1038/nsb0197-10. PMID 8989315.
- Wolynes P, Luthey-Schulten Z, Onuchic J (June 1996). "Fast-folding experiments and the topography of protein folding energy landscapes". Chemistry & Biology. 3 (6): 425–32. doi:10.1016/s1074-5521(96)90090-3. PMID 8807873.
- Dobson CM (December 2003). "Protein folding and misfolding". Nature. 426 (6968): 884–90. Bibcode:2003Natur.426..884D. doi:10.1038/nature02261. PMID 14685248.
- Onuchic JN, Wolynes PG (February 2004). "Theory of protein folding". Current Opinion in Structural Biology. 14 (1): 70–5. doi:10.1016/j.sbi.2004.01.009. PMID 15102452.
- Rollins GC, Dill KA (August 2014). "General mechanism of two-state protein folding kinetics". Journal of the American Chemical Society. 136 (32): 11420–7. doi:10.1021/ja5049434. PMC 5104671. PMID 25056406.
Further reading
- Dobson CM (2000-12-15). "The nature and significance of protein folding". In RH Pain (ed.). Mechanisms of Protein Folding (2nd ed.). Oxford, UK: Oxford University Press. ISBN 978-0-19-963788-1.
- Matagne A, Chung EW, Ball LJ, Radford SE, Robinson CV, Dobson CM (April 1998). "The origin of the alpha-domain intermediate in the folding of hen lysozyme". Journal of Molecular Biology. 277 (5): 997–1005. doi:10.1006/jmbi.1998.1657. PMID 9571017.