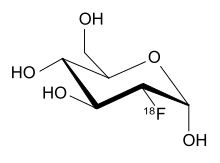
Fludeoxyglucose (18F)
Fluorodeoxyglucose (18F) (INN), or fluorodeoxyglucose F 18 (USAN and USP), also commonly called fluorodeoxyglucose and abbreviated [18F]FDG, 18F-FDG or FDG, is a radiopharmaceutical used in the medical imaging modality positron emission tomography (PET). Chemically, it is 2-deoxy-2-[18F]fluoro-D-glucose, a glucose analog, with the positron-emitting radionuclide fluorine-18 substituted for the normal hydroxyl group at the C-2 position in the glucose molecule.
 | |
| Names | |
|---|---|
| IUPAC name
2-Deoxy-2-[18F]fluoroglucose | |
| Identifiers | |
| |
3D model (JSmol) |
|
| Abbreviations | [18F]FDG |
| 2047723 | |
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| KEGG | |
PubChem CID |
|
| UNII | |
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| C6H1118FO5 | |
| Molar mass | 181.1495 g mol−1 |
| Melting point | 170 to 176[1] °C (338 to 349 °F; 443 to 449 K) |
| Pharmacology | |
| V09IX04 (WHO) | |
| Intravenous | |
| Pharmacokinetics: | |
| 6-Phosphorylation | |
| 110 min (at 70%) 16 min (at 20%) | |
| 20% Radioactivity renally excreted in two hours | |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
The uptake of 18F-FDG by tissues is a marker for the tissue uptake of glucose, which in turn is closely correlated with certain types of tissue metabolism. After 18F-FDG is injected into a patient, a PET scanner can form two-dimensional or three-dimensional images of the distribution of 18F-FDG within the body.
Since its development in 1976, 18F-FDG had a profound influence on research in the neurosciences.[2] The subsequent discovery in 1980 that 18F-FDG accumulates in tumors underpins the evolution of PET as a major clinical tool in cancer diagnosis.[3] 18F-FDG is now the standard radiotracer used for PET neuroimaging and cancer patient management.[4]
The images can be assessed by a nuclear medicine physician or radiologist to provide diagnoses of various medical conditions.
History
In 1968, Dr. Josef Pacák, Zdeněk Točík and Miloslav Černý at the Department of Organic Chemistry, Charles University, Czechoslovakia were the first to describe the synthesis of FDG.[5] Later, in the 1970s, Tatsuo Ido and Al Wolf at the Brookhaven National Laboratory were the first to describe the synthesis of FDG labeled with 18F.[6] The compound was first administered to two normal human volunteers by Abass Alavi in August, 1976 at the University of Pennsylvania. Brain images obtained with an ordinary (non-PET) nuclear scanner demonstrated the concentration of 18F-FDG in that organ (see history reference below).
Beginning in August 1990, and continuing throughout 1991, a shortage of oxygen-18, a raw material for FDG, made it necessary to ration isotope supplies. Israel's oxygen-18 facility had shut down due to the Gulf War, and the U.S. government had shut down its Isotopes of Carbon, Oxygen and Nitrogen facility at Los Alamos National Laboratory, leaving Isotec as the main supplier.[7]
Synthesis
[18F]FDG was first synthesized via electrophilic fluorination with [18F]F2.[6] Subsequently, a "nucleophilic synthesis" was devised with the same radioisotope.
As with all radioactive 18F-labeled radioligands, the 18F must be made initially as the fluoride anion in a cyclotron. Synthesis of complete [18F]FDG radioactive tracer begins with synthesis of the unattached fluoride radiotracer, since cyclotron bombardment destroys organic molecules of the type usually used for ligands, and in particular, would destroy glucose.
Cyclotron production of 18F may be accomplished by bombardment of neon-20 with deuterons, but usually is done by proton bombardment of 18O-enriched water, causing a (p,n) reaction (sometimes called a "knockout reaction" – a common type of nuclear reaction with high probability where an incoming proton "knocks out" a neutron) in the 18O. This produces "carrier-free" dissolved [18F]fluoride ([18F]F−) ions in the water. The 109.8-minute half-life of 18F makes rapid and automated chemistry necessary after this point.
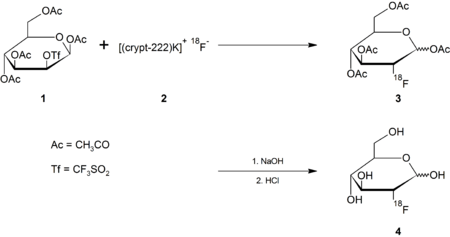
Anhydrous fluoride salts, which are easier to handle than fluorine gas, can be produced in a cyclotron.[8] To achieve this chemistry, the [18F]F− is separated from the aqueous solvent by trapping it on an ion-exchange column, and eluted with an acetonitrile solution of 2,2,2-cryptand and potassium carbonate. Evaporation of the eluate gives [(crypt-222)K]+ [18F]F− (2) .
The fluoride anion is nucleophilic but its anhydrous conditions are required to avoid competing reactions involving hydroxide, which is also a good nucleophile. The use of the cryptand to sequester the potassium ions avoids ion-pairing between free potassium and fluoride ions, rendering the fluoride anion more reactive.
Intermediate 2 is treated with the protected mannose triflate (1); the fluoride anion displaces the triflate leaving group in an SN2 reaction, giving the protected fluorinated deoxyglucose (3). Base hydrolysis removes the acetyl protecting groups, giving the desired product (4) after removing the cryptand via ion-exchange:[9][10]
Mechanism of action, metabolic end-products, and metabolic rate
[18F]FDG, as a glucose analog, is taken up by high-glucose-using cells such as brain, brown adipocytes, kidney, and cancer cells, where phosphorylation prevents the glucose from being released again from the cell, once it has been absorbed. The 2-hydroxyl group (–OH) in normal glucose is needed for further glycolysis (metabolism of glucose by splitting it), but [18F]FDG is missing this 2-hydroxyl. Thus, in common with its sister molecule 2-deoxy-D-glucose, FDG cannot be further metabolized in cells. The [18F]FDG-6-phosphate formed when [18F]FDG enters the cell thus cannot move out of the cell before radioactive decay. As a result, the distribution of [18F]FDG is a good reflection of the distribution of glucose uptake and phosphorylation by cells in the body.
After [18F]FDG decays radioactively, however, its 2-fluorine is converted to 18O−, and after picking up a proton H+ from a hydronium ion in its aqueous environment, the molecule becomes glucose-6-phosphate labeled with harmless nonradioactive "heavy oxygen" in the hydroxyl at the C-2 position. The new presence of a 2-hydroxyl now allows it to be metabolized normally in the same way as ordinary glucose, producing non-radioactive end-products.
Although in theory all [18F]FDG is metabolized as above with a radioactivity elimination half-life of 110 minutes (the same as that of fluorine-18), clinical studies have shown that the radioactivity of [18F]FDG partitions into two major fractions. About 75% of the fluorine-18 activity remains in tissues and is eliminated with a half-life of 110 minutes, presumably by decaying in place to O-18 to form [18O]O-glucose-6-phosphate, which is non-radioactive (this molecule can soon be metabolized to carbon dioxide and water, after nuclear transmutation of the fluorine to oxygen ceases to prevent metabolism). Another fraction of [18F]FDG, representing about 20% of the total fluorine-18 activity of an injection, is eliminated renally by two hours after a dose of [18F]FDG, with a rapid half-life of about 16 minutes (this portion makes the renal-collecting system and bladder prominent in a normal PET scan). This short biological half-life indicates that this 20% portion of the total fluorine-18 tracer activity is eliminated pharmacokinetically (through the renal system) much more quickly than the isotope itself can decay. The rapidity also suggests that some of this 18F is no longer attached to glucose, since low concentrations of glucose in the blood are retained by the normal kidney and not passed into the urine. Because of this rapidly excreted urine 18F, the urine of a patient undergoing a PET scan may therefore be especially radioactive for several hours after administration of the isotope.[11]
All radioactivity of [18F]FDG, both the 20% which is rapidly excreted in the first several hours of urine which is made after the exam, and the 80% which remains in the patient, decays with a half-life of 110 minutes (just under two hours). Thus, within 24 hours (13 half-lives after the injection), the radioactivity in the patient and in any initially voided urine which may have contaminated bedding or objects after the PET exam will have decayed to 2−13 = 1/8192 of the initial radioactivity of the dose. In practice, patients who have been injected with [18F]FDG are told to avoid the close vicinity of especially radiation-sensitive persons, such as infants, children and pregnant women, for at least 12 hours (7 half-lives, or decay to 1⁄128 the initial radioactive dose).
Distribution
The labeled 18F-FDG compound has a relatively short shelf life which is dominated by the physical decay of 18F with a half-life of 109.8 minutes, or slightly less than two hours. Still, this half life is sufficiently long to allow shipping the compound to remote PET scanning facilities, in contrast to other medical radioisotopes like 11C. Due to transport regulations for radioactive compounds, delivery is normally done by specially licensed road transport, but means of transport may also include dedicated small commercial jet services. Transport by air allows to expand the distribution area around a 18F-FDG production site to deliver the compound to PET scanning centres even hundreds of miles away.
Recently, on-site cyclotrons with integral shielding and portable chemistry stations for making 18F-FDG have accompanied PET scanners to remote hospitals. This technology holds some promise in the future, for replacing some of the scramble to transport 18F-FDG from site of manufacture to site of use.[12]
Production
Alliance Medical and Siemens Healthcare are the only producers in the United Kingdom. A dose of FDG in England costs about £130. In Northern Ireland, where there is a single supplier, doses cost up to £450.[13] IBA Molecular North America and Zevacor Molecular, both of which are owned by Illinois Health and Science (IBAM having been purchased as of 1 August 2015), Siemens' PETNET Solutions (a subsidiary of Siemens Healthcare), and Cardinal Health[14] are producers in the U.S.[15][16][17][18]
Applications
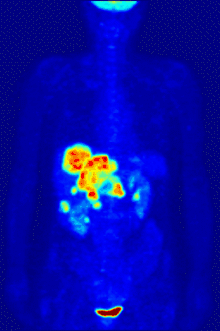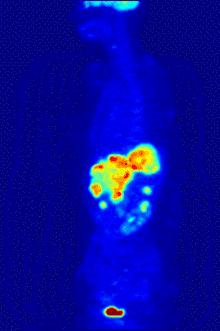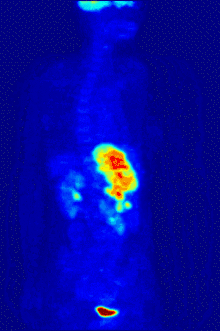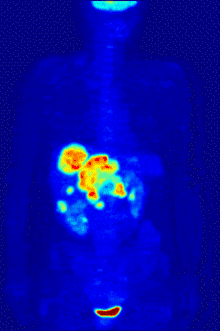
In PET imaging, 18F-FDG can be used for the assessment of glucose metabolism in the heart, lungs,[19] and the brain. It is also used for imaging tumors in oncology, where a static 18F-FDG PET scan is performed and the tumor 18F-FDG uptake is analyzed in terms of Standardized Uptake Value (SUV). 18F-FDG is taken up by cells, phosphorylated by hexokinase (whose mitochondrial form is greatly elevated in rapidly growing malignant tumours),[20] and retained by tissues with high metabolic activity, such as most types of malignant tumours. As a result, FDG-PET can be used for diagnosis, staging, and monitoring treatment of cancers, particularly in Hodgkin's disease, non-Hodgkin lymphoma, colorectal cancer, breast cancer, melanoma, and lung cancer. It has also been approved for use in diagnosing Alzheimer's disease.
In body-scanning applications in searching for tumor or metastatic disease, a dose of 18F-FDG in solution (typically 5 to 10 millicuries or 200 to 400 MBq) is typically injected rapidly into a saline drip running into a vein, in a patient who has been fasting for at least six hours, and who has a suitably low blood sugar. (This is a problem for some diabetics; usually PET scanning centers will not administer the isotope to patients with blood glucose levels over about 180 mg/dL = 10 mmol/L, and such patients must be rescheduled). The patient must then wait about an hour for the sugar to distribute and be taken up into organs which use glucose – a time during which physical activity must be kept to a minimum, in order to minimize uptake of the radioactive sugar into muscles (this causes unwanted artifacts in the scan, interfering with reading especially when the organs of interest are inside the body vs. inside the skull). Then, the patient is placed in the PET scanner for a series of one or more scans which may take from 20 minutes to as long as an hour (often, only about one-quarter of the body length may be imaged at a time).
See also
References
- Pacák J, Točík Z, Černý M (1969). "Synthesis of 2-Deoxy-2-fluoro-D-glucose". Journal of the Chemical Society D: Chemical Communications. 1969: 77–77. doi:10.1039/C29690000077.CS1 maint: multiple names: authors list (link)
- Newberg A, Alavi A, Reivich M (2002). "Determination of regional cerebral function with FDG-PET imaging in neuropsychiatric disorders". Semin Nucl Med. 32 (1): 13–14. doi:10.1053/snuc.2002.29276. PMID 11839066.
- Som P, Atkins HL, Bandoypadhyay D, Fowler JS, MacGregor RR, Matsui K, Oster ZH, Sacker DF, Shiue CY, Turner H, Wan CN, Wolf AP, Zabinski SV (1980). "A fluorinated glucose analog, 2-fluoro-2-deoxy-D-glucose (F-18): Nontoxic tracer for rapid tumor detection". J Nucl Med. 21 (7): 670–675. PMID 7391842.
- Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY, Cheson BD, O'shaughnessy J, Guyton KZ, Mankoff DA, Shankar L, Larson SM, Sigman CC, Schilsky RL, Sullivan DC (2005). "Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development". Clin. Cancer Res. 11 (8): 2785–2808. doi:10.1158/1078-0432.CCR-04-2626. PMID 15837727.
- Pacák J, Točík Z, Černý M (1969). "Synthesis of 2-Deoxy-2-fluoro-D-glucose". Journal of the Chemical Society D: Chemical Communications (2): 77. doi:10.1039/C29690000077.
- Ido T, Wan CN, Casella V, Fowler JS, Wolf AP, Reivich M, Kuhl DE (1978). "Labeled 2-deoxy-D-glucose analogs: 18F-labeled 2-deoxy-2-fluoro-D-glucose, 2-deoxy-2-fluoro-D-mannose and 14C-2-deoxy-2-fluoro-D-glucose". J Labeled Compounds Radiopharm. 24: 174–183. doi:10.1002/jlcr.2580140204.
- "Shortage of FDG raw material threatens expanded use of PET". DiagnosticImaging. 21 October 1992.
- Janet Miller, Radiopharmaceutical development at the Massachusetts General hospital (PDF), archived from the original (PDF) on 11 February 2015, retrieved 12 June 2013
- Fowler JS, Ido T (2002). "Initial and subsequent approach for the synthesis of 18FDG". Semin Nucl Med. 32 (1): 6–12. doi:10.1053/snuc.2002.29270. PMID 11839070.
- Yu, S (2006). "Review of 18F-FDG synthesis and quality control". Biomedical Imaging and Intervention Journal. 2 (4). doi:10.2349/biij.2.4.e57. PMC 3097819. PMID 21614337.
- "Fludeoxyglucose drug information". Retrieved 30 June 2009.
- Lisa Fratt (2003). "Radiation Testing and PET Minding the Radiopharmaceutical Store". Medical Imaging. Archived from the original on 20 November 2008.
- "'Monopoly' fears over £350m scans contract". Local Government Chronicle. 12 February 2015. Retrieved 22 February 2015.
- http://www.cardinalhealth.com/en/product-solutions/pharmaceutical-products/nuclear-medicine/safety-and-compliance/pet-biomarker-manufacturing.html
- "Fludeoxyglucose F 18- fludeoxyglucose f-18 injection". DailyMed. 8 May 2018. Retrieved 29 January 2020.
- "Fluorodeoxyglucose (F-18 FDG)". Archived from the original on 25 July 2015. Retrieved 24 July 2015.
- "National Drug Code Directory", U.S. Food and Drug Administration, Retrieved 5 February 2016.
- Matthews, Marianne (19 September 2013). "Siemens' PETNET Solutions to Aid The US Oncology Network". Axis Imaging News. Retrieved 5 February 2016.
- Gray's Anatomy for Students, Drake et al., 2005
- Ernesto Bustamante; Peter L. Pedersen (1977). "High Aerobic Glycolysis of Rat Hepatoma Cells in Culture: Role of Mitochondrial Hexokinase". Proceedings of the National Academy of Sciences. 74 (9): 3735–3739. Bibcode:1977PNAS...74.3735B. doi:10.1073/pnas.74.9.3735. PMC 431708. PMID 198801.
External links
- "Fludeoxyglucose F 18". Drug Information Portal. U.S. National Library of Medicine.