DMSO reductase
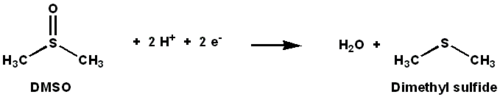
DMSO reductase is a molybdenum-containing enzyme that catalyzes reduction of dimethyl sulfoxide (DMSO) to dimethyl sulfide (DMS). This enzyme serves as the terminal reductase under anaerobic conditions in some bacteria, with DMSO being the terminal electron acceptor. During the course of the reaction, the oxygen atom in DMSO is transferred to molybdenum, and then reduced to water.
| Dimethylsulfoxide reductase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC number | 1.8.5.3 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| |||||||||
DMSO reductase (DMSOR) and other members of the DMSO reductase family are unique to bacteria and archaea. Enzymes of this family in anaerobic oxidative phosphorylation and inorganic-donor-based lithotrophic respiration. These enzymes have been engineered to degrade oxoanions.[1][2][3] DMSOR catalyzes the transfer of two electrons and one oxygen atom in the reaction: The active site of DMSOR contains molybdenum, which is otherwise rare in biology.[2]
Tertiary structure and active site
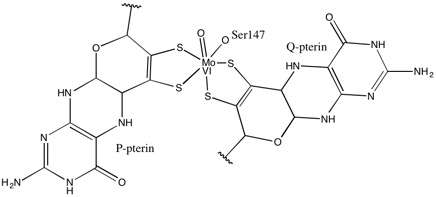
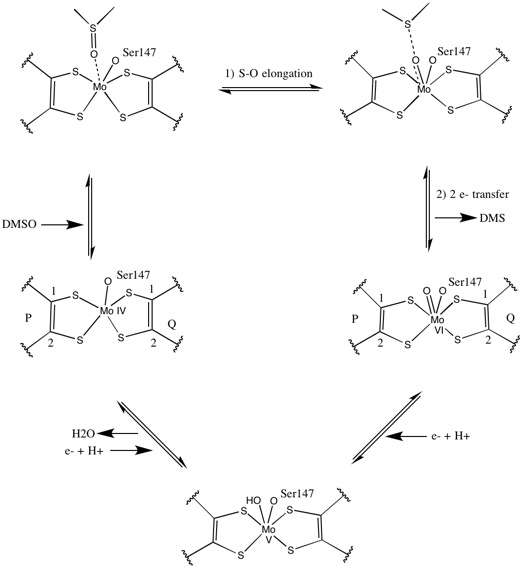
As for other members of DMSO reductase family, the tertiary structure of DMSOR is composed of Mo-surrounding domains I-IV, with domain IV heavily interacting with pyranopterindithiolene Mo-cofactor(s) (P- and Q-pterin) of the active site.[2][3] Members of the DMSO reductase family differ in terms of their active sites.[3] In the case of DMSOR, the Mo center is found to two dithiolene provided by two pyranopterin cofactors. These organic cofactors, called molybdopterins, are linked to GMP to create a dinucleotide form. An additional fifth cap-like ligand is the side-chain O of serine-147 residue, further classifying the enzyme as Type III DMSO reductase. InType I and II serine is replaced by cysteine and aspartate residues, respectively. Depending on the redox state of the Mo, which fluctuates between IV, V, or VI as the reaction progresses, the active site Mo core can also be ligated to an oxygen atom of an aqua-, hydroxo-, or oxo-group, respectively. Studies have shown that the particular identity of the amino-acid used to coordinate the Mo core greatly influences Mo redox midpoint potential and protonation state of the oxygen-group ligation, which are key determinants in the enzyme's mechanism for catalysis.[2]
Mechanism
Initial isotopic DMSO18 studies established a double-oxotransferase mechanism for DMSOR of R. sphaeroides. In this mechanism the labeled O18 is transferred from substrate to Mo, which then transfers the O18 to 1,3,5-triaza-7-phosphaadamantane (PTA) to yield PTAO18.[6] In an analogous mechanism, DMSO transfers O to Mo, and the resulting Mo(VI)O center is reduced, yielding water.[7]
Studies on synthetic Mo bis-dithiolene complexes suggest that be oxygen-transfer, electron transfer. Using S K-edge XAS and DFT, these model studies point to concerted S-O scission and electron transfer. Rates are proportion to decreasing substrate X-O bond strength and increasing substrate proton affinity.[8]
X-ray crystallography established that the overall tertiary structure of the enzyme remains constant through the reaction progression. However, several different experiments conducted on DMSOR of R. sphaeroides reported different results for the coordination activity of the four potential dithiolene ligands. While one x-ray crystallography investigation concluded equidistant coordination of all four Mo-S ligands in the oxidized form, which is supported by numerous x-ray absorption spectroscopy (XAS) studies, a different study characterized asymmetrical Mo-S distances. Both studies as well as electron paramagnetic resonance (EPR) studies have predicted that the Mo active site is highly flexible in terms of position and degree of potential ligand coordinations.[7][9]
The data that suggested two significantly asymmetric pyranopterin cofactors were used to propose a reaction mechanism. In the fully oxidized Mo VI form of the active site, the oxo-group and serine ligands were coordinated at 1.7 A distances from the Mo center. S1 and S2 of the P-pterin and S1 of the Q-pterin were locationed 2.4 A away from the Mo, and S2 of Q-pterin was located 3.1 A away. This pterin asymmetry may be the result of the trans-effect of the oxo-group weakening the S2-Mo bond, which is located directly opposite the oxo-group.[7]
In contrast, the structure of the fully reduced Mo IV form of the active site showed S1 and S2 P-pterin and S1 Q-pterin maintained full coordination, however the S2 of the Q-pterin shifted away from the metal center, indicating decreased coordination. This shift in ligand-Mo bond length is consistent with the proposed mechanism of direct oxygen transfer from the DMSO substrate to the Mo. A weaker dithiolene coordination in the reduced enzyme form could facilitate direct binding of the S=O. In the reduction of Mo and protonation of the oxo-group, it is proposed that a cytochrome electron source could bind to a depression above the active site and directly reduce the Mo center, or alternatively this cytochrome could bind to a well-solvated polypeptide loop in proximity to the Q-pterin, and Q-pterin could mediate this electron transfer.[7]
Cellular location and regulation
In R. sphaeroides, DMSOR is a single-subunit, water-soluble protein that requires no additional cofactors beyond pterin. In E. coli, DMSOR is embedded within the membrane and has three unique subunits, one of which includes the characteristic pterin cofactor, another which contains four 4Fe:4S clusters, and a final transmembrane subunit that binds and oxidizes menaquinol. The transfer of an e- from menaquinol to the 4Fe:4S clusters and finally to the pterin-Mo active site generates a proton gradient used for ATP generation.[7]
DMSOR regulated predominantly at a transcriptional level. It is encoded by the dor gene and expressed when activated by a signal cascade, which is under the regulation of DorS, DorR, and DorC proteins. A study of lacZ fusions (reporter genes) to corresponding dorS, dorR, and dorC promoters concluded that expression of DorR and DorC increased in reduced oxygen environments, but DorS expression was unaffected by oxygen concentration. DorC expression also increased with increasing concentrations of DMSO.[10]
Environmental impact
DMS, a product of DMSOR, is a component of the sulfur cycle. DMS is oxidized to Methanesulfonates, which nucleate cloud condensation over open oceans, where the alternative source of nucleation, dust, is absent. Cloud formation is a key component in increasing earth's albedo and regulating atmospheric temperature, thus this enzyme and the reaction it catalyzes could prove helpful on the climate control frontier.[11]
References
- Kappler U, Schäfer H (2014). "Chapter 11. Transformations of Dimethylsulfide". In Kroneck PM, Torres ME (eds.). The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment. Metal Ions in Life Sciences. 14. Springer. pp. 279–313. doi:10.1007/978-94-017-9269-1_11. ISBN 978-94-017-9268-4. PMID 25416398.
- McEwan AG, Kappler U (2004). "The DMSO Reductase Family of Microbial Molybdenum Enzymes" (PDF). Australian Biochemist. 35 (3): 17–20. Archived from the original (PDF) on 2014-03-07. Retrieved 2014-02-27.
- McEwan AG, Ridge JP, McDevitt CA, Hugenholtz P (2002). "The DMSO Reductase Family of Microbial Molybdenum Enzymes; Molecular Properties and Role in the Dissimilatory Reduction of Toxic Elements". Geomicrobiology Journal. 19 (1): 3–21. doi:10.1080/014904502317246138.
- PDB: 1DMS; Schneider F, Löwe J, Huber R, Schindelin H, Kisker C, Knäblein J (October 1996). "Crystal structure of dimethyl sulfoxide reductase from Rhodobacter capsulatus at 1.88 A resolution". Journal of Molecular Biology. 263 (1): 53–69. doi:10.1006/jmbi.1996.0555. PMID 8890912.
- PDB: 4DMR; McAlpine AS, McEwan AG, Bailey S (January 1998). "The high resolution crystal structure of DMSO reductase in complex with DMSO". Journal of Molecular Biology. 275 (4): 613–23. doi:10.1006/jmbi.1997.1513. PMID 9466935.
- Schultz BE, Hille R, Holm RH (1995), "Direct oxygen atom transfer in the mechanism of action of Rhodobacter sphaeroides dimethyl sulfoxide reductase", Journal of the American Chemical Society, 117 (2): 827–828, doi:10.1021/ja00107a031, ISSN 0002-7863
- Kisker C, Schindelin H, Rees DC (1997). "Molybdenum-cofactor-containing enzymes: structure and mechanism" (PDF). Annual Review of Biochemistry. 66: 233–67. doi:10.1146/annurev.biochem.66.1.233. PMID 9242907.
- Tenderholt AL, Wang JJ, Szilagyi RK, Holm RH, Hodgson KO, Hedman B, Solomon EI (June 2010). "Sulfur K-edge X-ray absorption spectroscopy and density functional calculations on Mo(IV) and Mo(VI)=O bis-dithiolenes: insights into the mechanism of oxo transfer in DMSO reductase and related functional analogues". Journal of the American Chemical Society. 132 (24): 8359–71. doi:10.1021/ja910369c. PMC 2907113. PMID 20499905.
- McAlpine AS, McEwan AG, Shaw AL, Bailey S (1997). "Molybdenum active centre of DMSO reductase from Rhodobacter capsulatus: crystal structure of the oxidised enzyme at 1.82-A resolution and the dithionite-reduced enzyme at 2.8-A resolution". JBIC. 2 (6): 690–701. doi:10.1007/s007750050185.
- Gunsalus RP (November 1992). "Control of electron flow in Escherichia coli: coordinated transcription of respiratory pathway genes". Journal of Bacteriology. 174 (22): 7069–74. PMC 207394. PMID 1331024.
- Sarkar B (21 March 2002). Heavy Metals In The Environment. CRC Press. p. 456. ISBN 978-0-8247-4475-5.