Cytochrome c oxidase subunit I
Cytochrome c oxidase I (COX1) also known as mitochondrially encoded cytochrome c oxidase I (MT-CO1) is a protein that in humans is encoded by the MT-CO1 gene.[5] In other eukaryotes, the gene is called COX1, CO1, or COI.[6] Cytochrome c oxidase I is the main subunit of the cytochrome c oxidase complex. Mutations in MT-CO1 have been associated with Leber's hereditary optic neuropathy (LHON), acquired idiopathic sideroblastic anemia, Complex IV deficiency, colorectal cancer, sensorineural deafness, and recurrent myoglobinuria.[7][8][9]
| Cytochrome c oxidase subunit I | |||||||||
|---|---|---|---|---|---|---|---|---|---|
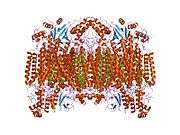
 Structure of the 13-subunit oxidized cytochrome c oxidase.[4] | |||||||||
| Identifiers | |||||||||
| Symbol | COX1 or COI | ||||||||
| Pfam | PF00115 | ||||||||
| InterPro | IPR000883 | ||||||||
| PROSITE | PDOC00074 | ||||||||
| SCOPe | 1occ / SUPFAM | ||||||||
| TCDB | 3.D.4 | ||||||||
| OPM superfamily | 4 | ||||||||
| OPM protein | 1v55 | ||||||||
| CDD | cd01663 | ||||||||
| |||||||||

Structure
One of 37 mitochondrial genes, the MT-CO1 gene is located from nucleotide pairs 5904 to 7444 on the guanine-rich heavy (H) section of mtDNA. The gene product is a 57 kDa protein composed of 513 amino acids.[10][11]
Function
Cytochrome c oxidase subunit I (CO1 or MT-CO1) is one of three mitochondrial DNA (mtDNA) encoded subunits (MT-CO1, MT-CO2, MT-CO3) of respiratory complex IV. Complex IV is the third and final enzyme of the electron transport chain of mitochondrial oxidative phosphorylation.[5]
Cytochrome c oxidase (EC 1.9.3.1) is a key enzyme in aerobic metabolism. Proton pumping heme-copper oxidases represent the terminal, energy-transfer enzymes of respiratory chains in prokaryotes and eukaryotes. The CuB-heme a3 (or heme o) binuclear centre, associated with the largest subunit I of cytochrome c and ubiquinol oxidases (EC 1.10.3.10), is directly involved in the coupling between dioxygen reduction and proton pumping.[12][13] Some terminal oxidases generate a transmembrane proton gradient across the plasma membrane (prokaryotes) or the mitochondrial inner membrane (eukaryotes).
The enzyme complex consists of 3-4 subunits (prokaryotes) up to 13 polypeptides (mammals) of which only the catalytic subunit (equivalent to mammalian subunit I (COI)) is found in all heme-copper respiratory oxidases. The presence of a bimetallic centre (formed by a high-spin heme and copper B) as well as a low-spin heme, both ligated to six conserved histidine residues near the outer side of four transmembrane spans within COI is common to all family members.[14][15][16] In contrast to eukaryotes the respiratory chain of prokaryotes is branched to multiple terminal oxidases. The enzyme complexes vary in heme and copper composition, substrate type and substrate affinity. The different respiratory oxidases allow the cells to customize their respiratory systems according to a variety of environmental growth conditions.[12]
It has been shown that eubacterial quinol oxidase was derived from cytochrome c oxidase in Gram-positive bacteria and that archaebacterial quinol oxidase has an independent origin. A considerable amount of evidence suggests that proteobacteria (Purple bacteria) acquired quinol oxidase through a lateral gene transfer from Gram-positive bacteria.[12]
A related nitric-oxide reductase (EC 1.7.99.7) exists in denitrifying species of archaea and eubacteria and is a heterodimer of cytochromes b and c. Phenazine methosulphate can act as acceptor. It has been suggested that cytochrome c oxidase catalytic subunits evolved from ancient nitric oxide reductases that could reduce both nitrogen and oxygen.[17][18]
Clinical significance
Mutations in this gene are associated with Leber's hereditary optic neuropathy (LHON), acquired idiopathic sideroblastic anemia, Complex IV deficiency, colorectal cancer, sensorineural deafness, and recurrent myoglobinuria.[7][8][9]
Leber's hereditary optic neuropathy (LHON)
LHON, correlated with mutations in MT-CO1, is characterized by optic nerve dysfunction, causing subacute or acute central vision loss. Some patients may display neurological or cardiac conduction defects. Because this disease is a result of mitochondrial DNA mutations affecting the respiratory chain complexes, it is inherited maternally.[19][8][9]
Acquired Idiopathic Sideroblastic Anemia
MT-CO1 may be involved in the development of acquired idiopathic sideroblastic anemia. Mutations in mitochondrial DNA can cause respiratory chain dysfunction, preventing reduction of ferric iron to ferrous iron, which is required for the final step in mitochondrial biosynthesis of heme. The result is a ferric accumulation in mitochondria and insufficient heme production.[20][21][8][9]
Mitochondrial Complex IV deficiency (MT-C4D)
Mutations in this gene can cause mitochondrial Complex IV deficiency, a disease of the mitochondrial respiratory chain displaying a wide variety of clinical manifestations ranging from isolated myopathy to a severe multisystem disease affecting multiple organs and tissues. Symptoms may include liver dysfunction and hepatomegaly, hypotonia, muscle weakness, exercise intolerance, delayed motor development, mental retardation, developmental delay, and hypertrophic cardiomyopathy. In some patients, the hypertrophic cardiomyopathy is fatal at the neonatal stage. Other affected individuals may manifest Leigh disease.[22][23][8][9]
Colorectal cancer (CRC)
MT-CO1 mutations play a role in colorectal cancer, a very complex disease displaying malignant lesions in the inner walls of the colon and rectum. Numerous such genetic alterations are often involved with the progression of adenoma, or premalignant lesions, to invasive adenocarcinoma. Long-standing ulcerative colitis, colon polyps, and family history are risk factors for colorectal cancer.[24][25][8][9]
Recurrent myoglobinuria mitochondrial (RM-MT)
RM-MT is a disease that is characterized by recurrent attacks of rhabdomyolysis (necrosis or disintegration of skeletal muscle) associated with muscle pain and weakness, exercise intolerance, low muscle capacity for oxidative phosphorylation, and followed by excretion of myoglobin in the urine. It has been associated with mitochondrial myopathy. A G5920A mutation, and a heteroplasmic G6708A nonsense mutation have been associated with COX deficiency and RM-MT.[26][27][8][9]
Deafness, sensorineural, mitochondrial (DFNM)
DFNM is a form of non-syndromic deafness with maternal inheritance. Affected individuals manifest progressive, postlingual, sensorineural hearing loss involving high frequencies. The mutation, A1555G, has been associated with this disease.[28][8][9]
Subfamilies
Use in DNA barcoding
MT-CO1 is a gene that is often used as a DNA barcode to identify animal species. The MT-CO1 gene sequence is suitable for this role because its mutation rate is often fast enough to distinguish closely related species and also because its sequence is conserved among conspecifics. Contrary to the primary objection raised by skeptics that MT-CO1 sequence differences are too small to be detected between closely related species, more than 2% sequence divergence is typically detected between closely related animal species,[29] suggesting that the barcode is effective for many animals. In most if not all seed plants, however, the rate of evolution of MT-CO1 is very slow.
MT-COI (= CCOI) in colonic crypts
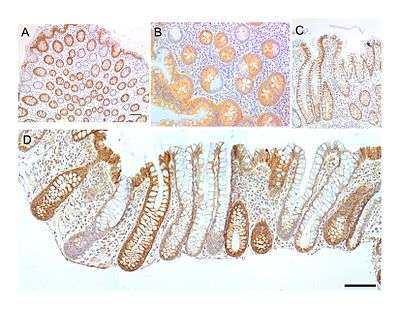
The MT-COI protein, also known as CCOI, is usually expressed at a high level in the cytoplasm of colonic crypts of the human large intestine (colon). However, MT-COI is frequently lost in colonic crypts with age in humans and is also often absent in field defects that give rise to colon cancers as well as in portions of colon cancers.[30]
The epithelial inner surface of the colon is punctuated by invaginations, the colonic crypts. The colon crypts are shaped like microscopic thick walled test tubes with a central hole down the length of the tube (the crypt lumen). Four tissue sections are shown in the image in this section, two cut across the long axes of the crypts and two cut parallel to the long axes.
Most of the human colonic crypts in the images have high expression of the brown-orange stained MT-COI. However, in some of the colonic crypts all of the cells lack MT-COI and appear mostly white, with their main color being the blue-gray staining of the nuclei at the outer walls of the crypts. Greaves et al.[31] showed that deficiencies of MT-COI in colonic crypts are due to mutations in the MT-COI gene. As seen in panel B, a portion of the stem cells of three crypts appear to have a mutation in MT-COI, so that 40% to 50% of the cells arising from those stem cells form a white segment in the cross-cut area.
In humans, the percent of colonic crypts deficient for MT-COI is less than 1% before age 40, but then increases linearly with age.[30] On average, the percent of colonic crypts deficient for MT-COI reaches 18% in women and 23% in men by 80–84 years of age.[30] Colonic tumors often arise in a field of crypts containing a large cluster (as many as 410) of MT-COI-deficient crypts. In colonic cancers, up to 80% of tumor cells can be deficient in MT-COI.[30]
As seen in panels C and D, crypts are about 75 to about 110 cells long. The average crypt circumference is 23 cells.[32] Based on these measurements, crypts have between 1725 and 2530 cells. Another report gave a range of 1500 to 4900 cells per colonic crypt.[33]
The occurrence of frequent crypts with almost complete loss of MT-COI in their 1700 to 5,000 cells suggests a process of natural selection. However, it has also been shown that a deficiency throughout a particular crypt due to an initial mitochondrial DNA mutation may occasionally occur through a stochastic process.[34][35] Nevertheless, the frequent occurrence of MT-COI deficiency in many crypts within a colon epithelium indicates that absence of MT-COI likely provides a selective advantage.
MT-COI is coded for by the mitochondrial chromosome. There are multiple copies of the chromosome in most mitochondria, usually between 2 and 6 per mitochondrion.[36][37][38] If a mutation occurs in MT-COI in one chromosome of a mitochondrion, there may be random segregation of the chromosomes during mitochondrial fission to generate new mitochondria. This can give rise to a mitochondrion with primarily or solely MT-COI-mutated chromosomes.
A mitochondrion with largely MT-COI-mutated chromosomes would need to have a positive selection bias in order to frequently become the main type of mitochondrion in a cell (a cell with MT-COI-deficient homoplasmy). There are about 100 to 700 mitochondria per cell, depending on cell type.[37][38] Furthermore, there is fairly rapid turnover of mitochondria, so that a mitochondrion with MT-COI-mutated chromosomes and a positive selection bias could shortly become the major type of mitochondrion in a cell. The average half-life of mitochondria in rats, depending on cell type, is between 9 and 24 days,[39] and in mice is about 2 days.[40] In humans it is likely that the half life of mitochondria is also a matter of days to weeks.
A stem cell at the base of a colonic crypt that was largely MT-COI-deficient may compete with the other 4 or 5 stem cells to take over the stem cell niche. If this occurs, then the colonic crypt would be deficient in MT-COI in all 1700 to 5,000 cells, as is indicated for some crypts in panels A, B and D of the image.
Crypts of the colon can reproduce by fission, as seen in panel C, where a crypt is fissioning to form two crypts, and in panel B where at least one crypt appears to be fissioning. Most crypts deficient in MT-COI are in clusters of crypts (clones of crypts) with two or more MT-COI-deficient crypts adjacent to each other (see panel D).[30] This illustrates that clones of deficient crypts often arise, and thus that there is likely a positive selective bias that has allowed them to spread in the human colonic epithelium.
It is not clear why a deficiency of MT-COI should have a positive selective bias. One suggestion[30] is that deficiency of MT-COI in a mitochondrion leads to lower reactive oxygen production (and less oxidative damage) and this provides a selective advantage in competition with other mitochondria within the same cell to generate homoplasmy for MT-COI-deficiency. Another suggestion was that cells with a deficiency in cytochrome c oxidase are apoptosis resistant, and thus more likely to survive. The linkage of MT-COI to apoptosis arises because active cytochrome c oxidase oxidizes cytochrome c, which then activates pro-caspase 9, leading to apoptosis.[41] These two factors may contribute to the frequent occurrence of MT-COI-deficient colonic crypts with age or during carcinogenesis in the human colon.
Interactions
Within the MITRAC (mitochondrial translation regulation assembly intermediate of cytochrome c oxidase) complex, the encoded protein interacts with COA3 and SMIM20/MITRAC7. This interaction with SMIM20 stabilizes the newly synthesized MT-CO1 and prevents its premature turnover.[42] Additionally, it interacts with TMEM177 in a COX20-dependent manner.[43][8][9]
References
- GRCm38: Ensembl release 89: ENSMUSG00000064351 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S (May 1996). "The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A". Science. 272 (5265): 1136–44. Bibcode:1996Sci...272.1136T. doi:10.1126/science.272.5265.1136. PMID 8638158.
- "Entrez Gene: Cytochrome c oxidase subunit I".
- Kosakyan A, Heger TJ, Leander BS, Todorov M, Mitchell EA, Lara E (May 2012). "COI barcoding of Nebelid testate amoebae (Amoebozoa: Arcellinida): extensive cryptic diversity and redefinition of the Hyalospheniidae Schultze" (PDF). Protist. 163 (3): 415–34. doi:10.1016/j.protis.2011.10.003. PMID 22130576.
- Online Mendelian Inheritance in Man (OMIM): 516030
- "MT-CO1 - Cytochrome c oxidase subunit 1 - Homo sapiens (Human) - MT-CO1 gene & protein". www.uniprot.org. Retrieved 2018-08-13.

- "UniProt: the universal protein knowledgebase". Nucleic Acids Research. 45 (D1): D158–D169. January 2017. doi:10.1093/nar/gkw1099. PMC 5210571. PMID 27899622.
- Zong NC, Li H, Li H, Lam MP, Jimenez RC, Kim CS, Deng N, Kim AK, Choi JH, Zelaya I, Liem D, Meyer D, Odeberg J, Fang C, Lu HJ, Xu T, Weiss J, Duan H, Uhlen M, Yates JR, Apweiler R, Ge J, Hermjakob H, Ping P (October 2013). "Integration of cardiac proteome biology and medicine by a specialized knowledgebase". Circulation Research. 113 (9): 1043–53. doi:10.1161/CIRCRESAHA.113.301151. PMC 4076475. PMID 23965338.
- "MT-CO1 - Cytochrome c oxidase subunit 1". Cardiac Organellar Protein Atlas Knowledgebase (COPaKB).
- García-Horsman JA, Barquera B, Rumbley J, Ma J, Gennis RB (September 1994). "The superfamily of heme-copper respiratory oxidases". Journal of Bacteriology. 176 (18): 5587–600. doi:10.1128/jb.176.18.5587-5600.1994. PMC 196760. PMID 8083153.
- Papa S, Capitanio N, Glaser P, Villani G (May 1994). "The proton pump of heme-copper oxidases". Cell Biology International. 18 (5): 345–55. doi:10.1006/cbir.1994.1084. PMID 8049679.
- Castresana J, Lübben M, Saraste M, Higgins DG (June 1994). "Evolution of cytochrome oxidase, an enzyme older than atmospheric oxygen". The EMBO Journal. 13 (11): 2516–25. doi:10.1002/j.1460-2075.1994.tb06541.x. PMC 395125. PMID 8013452.
- Capaldi RA, Malatesta F, Darley-Usmar VM (July 1983). "Structure of cytochrome c oxidase". Biochimica et Biophysica Acta (BBA) - Reviews on Bioenergetics. 726 (2): 135–48. doi:10.1016/0304-4173(83)90003-4. PMID 6307356.
- Holm L, Saraste M, Wikström M (September 1987). "Structural models of the redox centres in cytochrome oxidase". The EMBO Journal. 6 (9): 2819–23. doi:10.1002/j.1460-2075.1987.tb02578.x. PMC 553708. PMID 2824194.
- Saraste M, Castresana J (March 1994). "Cytochrome oxidase evolved by tinkering with denitrification enzymes". FEBS Letters. 341 (1): 1–4. doi:10.1016/0014-5793(94)80228-9. PMID 8137905.
- Chen J, Strous M (February 2013). "Denitrification and aerobic respiration, hybrid electron transport chains and co-evolution". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1827 (2): 136–44. doi:10.1016/j.bbabio.2012.10.002. PMID 23044391.
- Brown MD, Yang CC, Trounce I, Torroni A, Lott MT, Wallace DC (August 1992). "A mitochondrial DNA variant, identified in Leber hereditary optic neuropathy patients, which extends the amino acid sequence of cytochrome c oxidase subunit I". American Journal of Human Genetics. 51 (2): 378–85. PMC 1682694. PMID 1322638.
- Gattermann N, Retzlaff S, Wang YL, Hofhaus G, Heinisch J, Aul C, Schneider W (December 1997). "Heteroplasmic point mutations of mitochondrial DNA affecting subunit I of cytochrome c oxidase in two patients with acquired idiopathic sideroblastic anemia". Blood. 90 (12): 4961–72. doi:10.1182/blood.V90.12.4961. PMID 9389715.
- Bröker S, Meunier B, Rich P, Gattermann N, Hofhaus G (November 1998). "MtDNA mutations associated with sideroblastic anaemia cause a defect of mitochondrial cytochrome c oxidase". European Journal of Biochemistry. 258 (1): 132–8. doi:10.1046/j.1432-1327.1998.2580132.x. PMID 9851701.
- Varlamov DA, Kudin AP, Vielhaber S, Schröder R, Sassen R, Becker A, Kunz D, Haug K, Rebstock J, Heils A, Elger CE, Kunz WS (August 2002). "Metabolic consequences of a novel missense mutation of the mtDNA CO I gene". Human Molecular Genetics. 11 (16): 1797–805. doi:10.1093/hmg/11.16.1797. PMID 12140182.
- Lucioli S, Hoffmeier K, Carrozzo R, Tessa A, Ludwig B, Santorelli FM (March 2006). "Introducing a novel human mtDNA mutation into the Paracoccus denitrificans COX I gene explains functional deficits in a patient". Neurogenetics. 7 (1): 51–7. doi:10.1007/s10048-005-0015-z. PMID 16284789.
- Greaves LC, Preston SL, Tadrous PJ, Taylor RW, Barron MJ, Oukrif D, Leedham SJ, Deheragoda M, Sasieni P, Novelli MR, Jankowski JA, Turnbull DM, Wright NA, McDonald SA (January 2006). "Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission". Proceedings of the National Academy of Sciences of the United States of America. 103 (3): 714–9. Bibcode:2006PNAS..103..714G. doi:10.1073/pnas.0505903103. PMC 1325106. PMID 16407113.
- Namslauer I, Brzezinski P (March 2009). "A mitochondrial DNA mutation linked to colon cancer results in proton leaks in cytochrome c oxidase". Proceedings of the National Academy of Sciences of the United States of America. 106 (9): 3402–7. Bibcode:2009PNAS..106.3402N. doi:10.1073/pnas.0811450106. PMC 2651238. PMID 19218458.
- Karadimas CL, Greenstein P, Sue CM, Joseph JT, Tanji K, Haller RG, Taivassalo T, Davidson MM, Shanske S, Bonilla E, DiMauro S (September 2000). "Recurrent myoglobinuria due to a nonsense mutation in the COX I gene of mitochondrial DNA". Neurology. 55 (5): 644–9. doi:10.1212/wnl.55.5.644. PMID 10980727.
- Kollberg G, Moslemi AR, Lindberg C, Holme E, Oldfors A (February 2005). "Mitochondrial myopathy and rhabdomyolysis associated with a novel nonsense mutation in the gene encoding cytochrome c oxidase subunit I". Journal of Neuropathology and Experimental Neurology. 64 (2): 123–8. doi:10.1093/jnen/64.2.123. PMID 15751226.
- Pandya A, Xia XJ, Erdenetungalag R, Amendola M, Landa B, Radnaabazar J, Dangaasuren B, Van Tuyle G, Nance WE (December 1999). "Heterogenous point mutations in the mitochondrial tRNA Ser(UCN) precursor coexisting with the A1555G mutation in deaf students from Mongolia". American Journal of Human Genetics. 65 (6): 1803–6. doi:10.1086/302658. PMC 1288397. PMID 10577941.
- Hebert PD, Ratnasingham S, deWaard JR (August 2003). "Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species". Proceedings. Biological Sciences. 270 Suppl 1: S96–9. doi:10.1098/rsbl.2003.0025. PMC 1698023. PMID 12952648.
- Bernstein C, Facista A, Nguyen H, Zaitlin B, Hassounah N, Loustaunau C, Payne CM, Banerjee B, Goldschmid S, Tsikitis VL, Krouse R, Bernstein H (December 2010). "Cancer and age related colonic crypt deficiencies in cytochrome c oxidase I". World Journal of Gastrointestinal Oncology. 2 (12): 429–42. doi:10.4251/wjgo.v2.i12.429. PMC 3011097. PMID 21191537.
- Greaves LC, Preston SL, Tadrous PJ, Taylor RW, Barron MJ, Oukrif D, Leedham SJ, Deheragoda M, Sasieni P, Novelli MR, Jankowski JA, Turnbull DM, Wright NA, McDonald SA (January 2006). "Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission". Proceedings of the National Academy of Sciences of the United States of America. 103 (3): 714–9. Bibcode:2006PNAS..103..714G. doi:10.1073/pnas.0505903103. PMC 1325106. PMID 16407113.
- Baker AM, Cereser B, Melton S, Fletcher AG, Rodriguez-Justo M, Tadrous PJ, Humphries A, Elia G, McDonald SA, Wright NA, Simons BD, Jansen M, Graham TA (August 2014). "Quantification of crypt and stem cell evolution in the normal and neoplastic human colon". Cell Reports. 8 (4): 940–7. doi:10.1016/j.celrep.2014.07.019. PMC 4471679. PMID 25127143.
- Nooteboom M, Johnson R, Taylor RW, Wright NA, Lightowlers RN, Kirkwood TB, Mathers JC, Turnbull DM, Greaves LC (February 2010). "Age-associated mitochondrial DNA mutations lead to small but significant changes in cell proliferation and apoptosis in human colonic crypts". Aging Cell. 9 (1): 96–9. doi:10.1111/j.1474-9726.2009.00531.x. PMC 2816353. PMID 19878146.
- Coller HA, Bodyak ND, Khrapko K (April 2002). "Frequent intracellular clonal expansions of somatic mtDNA mutations: significance and mechanisms". Annals of the New York Academy of Sciences. 959 (1): 434–47. Bibcode:2002NYASA.959..434C. doi:10.1111/j.1749-6632.2002.tb02113.x. PMID 11976216.
- Nekhaeva E, Bodyak ND, Kraytsberg Y, McGrath SB, Van Orsouw NJ, Pluzhnikov A, Wei JY, Vijg J, Khrapko K (April 2002). "Clonally expanded mtDNA point mutations are abundant in individual cells of human tissues". Proceedings of the National Academy of Sciences of the United States of America. 99 (8): 5521–6. Bibcode:2002PNAS...99.5521N. doi:10.1073/pnas.072670199. PMC 122802. PMID 11943860.
- Legros F, Malka F, Frachon P, Lombès A, Rojo M (June 2004). "Organization and dynamics of human mitochondrial DNA". Journal of Cell Science. 117 (Pt 13): 2653–62. doi:10.1242/jcs.01134. PMID 15138283.
- Robin ED, Wong R (September 1988). "Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells". Journal of Cellular Physiology. 136 (3): 507–13. doi:10.1002/jcp.1041360316. PMID 3170646.
- Satoh M, Kuroiwa T (September 1991). "Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell". Experimental Cell Research. 196 (1): 137–40. doi:10.1016/0014-4827(91)90467-9. PMID 1715276.
- Menzies RA, Gold PH (April 1971). "The turnover of mitochondria in a variety of tissues of young adult and aged rats". The Journal of Biological Chemistry. 246 (8): 2425–9. PMID 5553400.
- Miwa S, Lawless C, von Zglinicki T (December 2008). "Mitochondrial turnover in liver is fast in vivo and is accelerated by dietary restriction: application of a simple dynamic model". Aging Cell. 7 (6): 920–3. doi:10.1111/j.1474-9726.2008.00426.x. PMC 2659384. PMID 18691181.
- Brown GC, Borutaite V (2008). "Regulation of apoptosis by the redox state of cytochrome c". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1777 (7–8): 877–81. doi:10.1016/j.bbabio.2008.03.024. PMID 18439415.
- Dennerlein S, Oeljeklaus S, Jans D, Hellwig C, Bareth B, Jakobs S, Deckers M, Warscheid B, Rehling P (September 2015). "MITRAC7 Acts as a COX1-Specific Chaperone and Reveals a Checkpoint during Cytochrome c Oxidase Assembly". Cell Reports. 12 (10): 1644–55. doi:10.1016/j.celrep.2015.08.009. PMID 26321642.
- Lorenzi I, Oeljeklaus S, Aich A, Ronsör C, Callegari S, Dudek J, Warscheid B, Dennerlein S, Rehling P (February 2018). "The mitochondrial TMEM177 associates with COX20 during COX2 biogenesis". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1865 (2): 323–333. doi:10.1016/j.bbamcr.2017.11.010. PMC 5764226. PMID 29154948.
Further reading
- Torroni A, Achilli A, Macaulay V, Richards M, Bandelt HJ (June 2006). "Harvesting the fruit of the human mtDNA tree". Trends in Genetics. 22 (6): 339–45. doi:10.1016/j.tig.2006.04.001. PMID 16678300.
- Bodenteich A, Mitchell LG, Polymeropoulos MH, Merril CR (May 1992). "Dinucleotide repeat in the human mitochondrial D-loop". Human Molecular Genetics. 1 (2): 140. doi:10.1093/hmg/1.2.140-a. PMID 1301157.
- Brown MD, Yang CC, Trounce I, Torroni A, Lott MT, Wallace DC (August 1992). "A mitochondrial DNA variant, identified in Leber hereditary optic neuropathy patients, which extends the amino acid sequence of cytochrome c oxidase subunit I". American Journal of Human Genetics. 51 (2): 378–85. PMC 1682694. PMID 1322638.
- Lu X, Walker T, MacManus JP, Seligy VL (July 1992). "Differentiation of HT-29 human colonic adenocarcinoma cells correlates with increased expression of mitochondrial RNA: effects of trehalose on cell growth and maturation". Cancer Research. 52 (13): 3718–25. PMID 1377597.
- Marzuki S, Noer AS, Lertrit P, Thyagarajan D, Kapsa R, Utthanaphol P, Byrne E (December 1991). "Normal variants of human mitochondrial DNA and translation products: the building of a reference data base". Human Genetics. 88 (2): 139–45. doi:10.1007/bf00206061. PMID 1757091.
- Moraes CT, Andreetta F, Bonilla E, Shanske S, DiMauro S, Schon EA (March 1991). "Replication-competent human mitochondrial DNA lacking the heavy-strand promoter region". Molecular and Cellular Biology. 11 (3): 1631–7. doi:10.1128/MCB.11.3.1631. PMC 369459. PMID 1996112.
- Attardi G, Chomyn A, Doolittle RF, Mariottini P, Ragan CI (1987). "Seven unidentified reading frames of human mitochondrial DNA encode subunits of the respiratory chain NADH dehydrogenase". Cold Spring Harbor Symposia on Quantitative Biology. 51 (1): 103–14. doi:10.1101/sqb.1986.051.01.013. PMID 3472707.
- Chomyn A, Cleeter MW, Ragan CI, Riley M, Doolittle RF, Attardi G (October 1986). "URF6, last unidentified reading frame of human mtDNA, codes for an NADH dehydrogenase subunit". Science. 234 (4776): 614–8. Bibcode:1986Sci...234..614C. doi:10.1126/science.3764430. PMID 3764430.
- Chomyn A, Mariottini P, Cleeter MW, Ragan CI, Matsuno-Yagi A, Hatefi Y, Doolittle RF, Attardi G (1985). "Six unidentified reading frames of human mitochondrial DNA encode components of the respiratory-chain NADH dehydrogenase". Nature. 314 (6012): 592–7. Bibcode:1985Natur.314..592C. doi:10.1038/314592a0. PMID 3921850.
- Sanger F, Coulson AR, Barrell BG, Smith AJ, Roe BA (October 1980). "Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing". Journal of Molecular Biology. 143 (2): 161–78. doi:10.1016/0022-2836(80)90196-5. PMID 6260957.
- Montoya J, Ojala D, Attardi G (April 1981). "Distinctive features of the 5'-terminal sequences of the human mitochondrial mRNAs". Nature. 290 (5806): 465–70. Bibcode:1981Natur.290..465M. doi:10.1038/290465a0. PMID 7219535.
- Horai S, Hayasaka K, Kondo R, Tsugane K, Takahata N (January 1995). "Recent African origin of modern humans revealed by complete sequences of hominoid mitochondrial DNAs". Proceedings of the National Academy of Sciences of the United States of America. 92 (2): 532–6. Bibcode:1995PNAS...92..532H. doi:10.1073/pnas.92.2.532. PMC 42775. PMID 7530363.
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (October 1999). "Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA". Nature Genetics. 23 (2): 147. doi:10.1038/13779. PMID 10508508.
- Ingman M, Kaessmann H, Pääbo S, Gyllensten U (December 2000). "Mitochondrial genome variation and the origin of modern humans". Nature. 408 (6813): 708–13. Bibcode:2000Natur.408..708I. doi:10.1038/35047064. PMID 11130070.
- Finnilä S, Lehtonen MS, Majamaa K (June 2001). "Phylogenetic network for European mtDNA". American Journal of Human Genetics. 68 (6): 1475–84. doi:10.1086/320591. PMC 1226134. PMID 11349229.
- Maca-Meyer N, González AM, Larruga JM, Flores C, Cabrera VM (2003). "Major genomic mitochondrial lineages delineate early human expansions". BMC Genetics. 2: 13. doi:10.1186/1471-2156-2-13. PMC 55343. PMID 11553319.
PDB gallery | |
|---|---|
|










This article incorporates text from the United States National Library of Medicine, which is in the public domain.