Cell cycle
The cell cycle, or cell-division cycle, is the series of events that take place in a cell that cause it to divide into two daughter cells. These events include the duplication of its DNA (DNA replication) and some of its organelles, and subsequently the partitioning of its cytoplasm and other components into two daughter cells in a process called cell division.
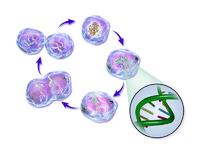
In cells with nuclei (eukaryotes), (i.e., animal, plant, fungal, and protist cells), the cell cycle is divided into two main stages: interphase and the mitotic (M) phase (including mitosis and cytokinesis). During interphase, the cell grows, accumulating nutrients needed for mitosis, and replicates its DNA and some of its organelles. During the mitotic phase, the replicated chromosomes, organelles, and cytoplasm separate into two new daughter cells. To ensure the proper replication of cellular components and division, there are control mechanisms known as cell cycle checkpoints after each of the key steps of the cycle that determine if the cell can progress to the next phase.
In cells without nuclei (prokaryotes), (i.e., bacteria and archaea), the cell cycle is divided into the B, C, and D periods. The B period extends from the end of cell division to the beginning of DNA replication. DNA replication occurs during the C period. The D period refers to the stage between the end of DNA replication and the splitting of the bacterial cell into two daughter cells.[1]
The cell-division cycle is a vital process by which a single-celled fertilized egg develops into a mature organism, as well as the process by which hair, skin, blood cells, and some internal organs are renewed. After cell division, each of the daughter cells begin the interphase of a new cycle. Although the various stages of interphase are not usually morphologically distinguishable, each phase of the cell cycle has a distinct set of specialized biochemical processes that prepare the cell for initiation of the cell division.
Phases
The eukaryotic cell cycle consists of four distinct phases: G1 phase, S phase (synthesis), G2 phase (collectively known as interphase) and M phase (mitosis and cytokinesis). M phase is itself composed of two tightly coupled processes: mitosis, in which the cell's nucleus divides, and cytokinesis, in which the cell's cytoplasm divides forming two daughter cells. Activation of each phase is dependent on the proper progression and completion of the previous one. Cells that have temporarily or reversibly stopped dividing are said to have entered a state of quiescence called G0 phase.
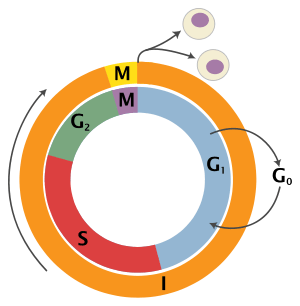
| State | Phase | Abbreviation | Description |
|---|---|---|---|
| Resting | Gap 0 | G0 | A phase where the cell has left the cycle and has stopped dividing. |
| Interphase | Gap 1 | G1 | Cells increase in size in Gap 1. The G1 checkpoint control mechanism ensures that everything is ready for DNA synthesis. |
| Synthesis | S | DNA replication occurs during this phase. | |
| Gap 2 | G2 | During the gap between DNA synthesis and mitosis, the cell will continue to grow. The G2 checkpoint control mechanism ensures that everything is ready to enter the M (mitosis) phase and divide. | |
| Cell division | Mitosis | M | Cell growth stops at this stage and cellular energy is focused on the orderly division into two daughter cells. A checkpoint in the middle of mitosis (Metaphase Checkpoint) ensures that the cell is ready to complete cell division. |
After cell division, each of the daughter cells begin the interphase of a new cycle. Although the various stages of interphase are not usually morphologically distinguishable, each phase of the cell cycle has a distinct set of specialized biochemical processes that prepare the cell for initiation of cell division.
G0 phase (quiescence)

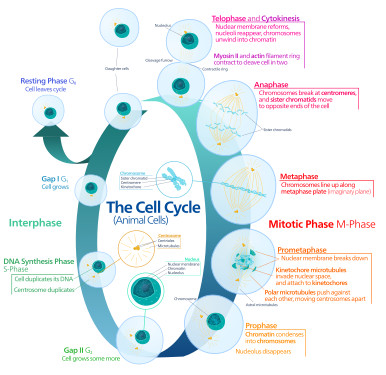
G0 is a resting phase where the cell has left the cycle and has stopped dividing. The cell cycle starts with this phase. Non-proliferative (non-dividing) cells in multicellular eukaryotes generally enter the quiescent G0 state from G1 and may remain quiescent for long periods of time, possibly indefinitely (as is often the case for neurons). This is very common for cells that are fully differentiated. Some cells enter the G0 phase semi-permanently and are considered post-mitotic, e.g., some liver, kidney, and stomach cells. Many cells do not enter G0 and continue to divide throughout an organism's life, e.g., epithelial cells.
The word "post-mitotic" is sometimes used to refer to both quiescent and senescent cells. Cellular senescence occurs in response to DNA damage and external stress and usually constitutes an arrest in G1. Cellular senescence may make a cell's progeny nonviable; it is often a biochemical alternative to the self-destruction of such a damaged cell by apoptosis.
Interphase
Interphase is a series of changes that takes place in a newly formed cell and its nucleus before it becomes capable of division again. It is also called preparatory phase or intermitosis. Typically interphase lasts for at least 91% of the total time required for the cell cycle.
Interphase proceeds in three stages, G1, S, and G2, followed by the cycle of mitosis and cytokinesis. The cell's nuclear DNA contents are duplicated during S phase.
G1 phase (First growth phase or Post mitotic gap phase)
The first phase within interphase, from the end of the previous M phase until the beginning of DNA synthesis, is called G1 (G indicating gap). It is also called the growth phase. During this phase, the biosynthetic activities of the cell, which are considerably slowed down during M phase, resume at a high rate. The duration of G1 is highly variable, even among different cells of the same species.[3] In this phase, the cell increases its supply of proteins, increases the number of organelles (such as mitochondria, ribosomes), and grows in size. In G1 phase, a cell has three options.
- To continue cell cycle and enter S phase
- Stop cell cycle and enter G0 phase for undergoing differentiation.
- Become arrested in G1 phase hence it may enter G0 phase or re-enter cell cycle.
The deciding point is called check point (Restriction point). This check point is called the restriction point or START and is regulated by G1/S cyclins, which cause transition from G1 to S phase. Passage through the G1 check point commits the cell to division.
S phase (DNA replication)
The ensuing S phase starts when DNA synthesis commences; when it is complete, all of the chromosomes have been replicated, i.e., each chromosome consists of two sister chromatids. Thus, during this phase, the amount of DNA in the cell has doubled, though the ploidy and number of chromosomes are unchanged. Rates of RNA transcription and protein synthesis are very low during this phase. An exception to this is histone production, most of which occurs during the S phase.[4][5][6]
G2 phase (growth)
G2 phase occurs after DNA replication and is a period of protein synthesis and rapid cell growth to prepare the cell for mitosis. During this phase microtubules begin to reorganize to form a spindle (preprophase). Before proceeding to mitotic phase, cells must be checked at the G2checkpoint for any DNA damage within the chromosomes. The G2 checkpoint is mainly regulated by the tumor protein p53. If the DNA is damaged, p53 will either repair the DNA or trigger the apoptosis of the cell. If p53 is dysfunctional or mutated, cells with damaged DNA may continue through the cell cycle, leading to the development of cancer.
Mitotic phase (chromosome separation)
The relatively brief M phase consists of nuclear division (karyokinesis). It is a relatively short period of the cell cycle. M phase is complex and highly regulated. The sequence of events is divided into phases, corresponding to the completion of one set of activities and the start of the next. These phases are sequentially known as:
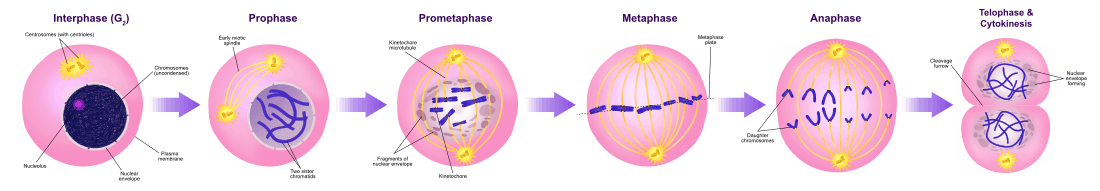
Mitosis is the process by which a eukaryotic cell separates the chromosomes in its cell nucleus into two identical sets in two nuclei.[7] During the process of mitosis the pairs of chromosomes condense and attach to microtubules that pull the sister chromatids to opposite sides of the cell.[8]
Mitosis occurs exclusively in eukaryotic cells, but occurs in different ways in different species. For example, animal cells undergo an "open" mitosis, where the nuclear envelope breaks down before the chromosomes separate, while fungi such as Aspergillus nidulans and Saccharomyces cerevisiae (yeast) undergo a "closed" mitosis, where chromosomes divide within an intact cell nucleus.[9]
Cytokinesis phase (separation of all cell components)
Mitosis is immediately followed by cytokinesis, which divides the nuclei, cytoplasm, organelles and cell membrane into two cells containing roughly equal shares of these cellular components. Mitosis and cytokinesis together define the division of the mother cell into two daughter cells, genetically identical to each other and to their parent cell. This accounts for approximately 10% of the cell cycle.
Because cytokinesis usually occurs in conjunction with mitosis, "mitosis" is often used interchangeably with "M phase". However, there are many cells where mitosis and cytokinesis occur separately, forming single cells with multiple nuclei in a process called endoreplication. This occurs most notably among the fungi and slime molds, but is found in various groups. Even in animals, cytokinesis and mitosis may occur independently, for instance during certain stages of fruit fly embryonic development.[10] Errors in mitosis can result in cell death through apoptosis or cause mutations that may lead to cancer.
Regulation of eukaryotic cell cycle
Regulation of the cell cycle involves processes crucial to the survival of a cell, including the detection and repair of genetic damage as well as the prevention of uncontrolled cell division. The molecular events that control the cell cycle are ordered and directional; that is, each process occurs in a sequential fashion and it is impossible to "reverse" the cycle.
Role of cyclins and CDKs
 Nobel Laureate Paul Nurse |
 Nobel Laureate Tim Hunt |
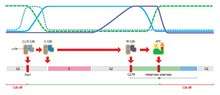
Two key classes of regulatory molecules, cyclins and cyclin-dependent kinases (CDKs), determine a cell's progress through the cell cycle.[11] Leland H. Hartwell, R. Timothy Hunt, and Paul M. Nurse won the 2001 Nobel Prize in Physiology or Medicine for their discovery of these central molecules.[12] Many of the genes encoding cyclins and CDKs are conserved among all eukaryotes, but in general more complex organisms have more elaborate cell cycle control systems that incorporate more individual components. Many of the relevant genes were first identified by studying yeast, especially Saccharomyces cerevisiae;[13] genetic nomenclature in yeast dubs many of these genes cdc (for "cell division cycle") followed by an identifying number, e.g. cdc25 or cdc20.
Cyclins form the regulatory subunits and CDKs the catalytic subunits of an activated heterodimer; cyclins have no catalytic activity and CDKs are inactive in the absence of a partner cyclin. When activated by a bound cyclin, CDKs perform a common biochemical reaction called phosphorylation that activates or inactivates target proteins to orchestrate coordinated entry into the next phase of the cell cycle. Different cyclin-CDK combinations determine the downstream proteins targeted. CDKs are constitutively expressed in cells whereas cyclins are synthesised at specific stages of the cell cycle, in response to various molecular signals.[14]
General mechanism of cyclin-CDK interaction
Upon receiving a pro-mitotic extracellular signal, G1 cyclin-CDK complexes become active to prepare the cell for S phase, promoting the expression of transcription factors that in turn promote the expression of S cyclins and of enzymes required for DNA replication. The G1 cyclin-CDK complexes also promote the degradation of molecules that function as S phase inhibitors by targeting them for ubiquitination. Once a protein has been ubiquitinated, it is targeted for proteolytic degradation by the proteasome. However, results from a recent study of E2F transcriptional dynamics at the single-cell level argue that the role of G1 cyclin-CDK activities, in particular cyclin D-CDK4/6, is to tune the timing rather than the commitment of cell cycle entry.[15]
Active S cyclin-CDK complexes phosphorylate proteins that make up the pre-replication complexes assembled during G1 phase on DNA replication origins. The phosphorylation serves two purposes: to activate each already-assembled pre-replication complex, and to prevent new complexes from forming. This ensures that every portion of the cell's genome will be replicated once and only once. The reason for prevention of gaps in replication is fairly clear, because daughter cells that are missing all or part of crucial genes will die. However, for reasons related to gene copy number effects, possession of extra copies of certain genes is also deleterious to the daughter cells.
Mitotic cyclin-CDK complexes, which are synthesized but inactivated during S and G2 phases, promote the initiation of mitosis by stimulating downstream proteins involved in chromosome condensation and mitotic spindle assembly. A critical complex activated during this process is a ubiquitin ligase known as the anaphase-promoting complex (APC), which promotes degradation of structural proteins associated with the chromosomal kinetochore. APC also targets the mitotic cyclins for degradation, ensuring that telophase and cytokinesis can proceed.[16]
Specific action of cyclin-CDK complexes
Cyclin D is the first cyclin produced in the cells that enter the cell cycle, in response to extracellular signals (e.g. growth factors). Cyclin D levels stay low in resting cells that are not proliferating. Additionally, CDK4/6 and CDK2 are also inactive because CDK4/6 are bound by INK4 family members (e.g., p16), limiting kinase activity. Meanwhile, CDK2 complexes are inhibited by the CIP/KIP proteins such as p21 and p27,[17] When it is time for a cell to enter the cell cycle, which is triggered by a mitogenic stimuli, levels of cyclin D increase. In response to this trigger, cyclin D binds to existing CDK4/6, forming the active cyclin D-CDK4/6 complex. Cyclin D-CDK4/6 complexes in turn mono-phosphorylates the retinoblastoma susceptibility protein (Rb) to pRb. The un-phosphorylated Rb tumour suppressor functions in inducing cell cycle exit and maintaining G0 arrest (senescence).[18]
In the last few decades, a model has been widely accepted whereby pRB proteins are inactivated by cyclin D-Cdk4/6-mediated phosphorylation. Rb has 14+ potential phosphorylation sites. Cyclin D-Cdk 4/6 progressively phosphorylates Rb to hyperphosphorylated state, which triggers dissociation of pRB–E2F complexes, thereby inducing G1/S cell cycle gene expression and progression into S phase.[19]
However, scientific observations from a recent study show that Rb is present in three types of isoforms: (1) un-phosphorylated Rb in G0 state; (2) mono-phosphorylated Rb, also referred to as “hypo-phosphorylated’ or ‘partially’ phosphorylated Rb in early G1 state; and (3) inactive hyper-phosphorylated Rb in late G1 state.[20][21][22] In early G1 cells, mono-phosphorylated Rb exits as 14 different isoforms, one of each has distinct E2F binding affinity.[22] Rb has been found to associate with hundreds of different proteins [23] and the idea that different mono-phosphorylated Rb isoforms have different protein partners was very appealing.[24] A recent report confirmed that mono-phosphorylation controls Rb's association with other proteins and generates functional distinct forms of Rb.[25] All different mono-phosphorylated Rb isoforms inhibit E2F transcriptional program and are able to arrest cells in G1-phase. Importantly, different mono-phosphorylated forms of RB have distinct transcriptional outputs that are extended beyond E2F regulation.[25]
In general, the binding of pRb to E2F inhibits the E2F target gene expression of certain G1/S and S transition genes including E-type cyclins. The partial phosphorylation of RB de-represses the Rb-mediated suppression of E2F target gene expression, begins the expression of cyclin E. The molecular mechanism that causes the cell switched to cyclin E activation is currently not known, but as cyclin E levels rise, the active cyclin E-CDK2 complex is formed, bringing Rb to be inactivated by hyper-phosphorylation.[22] Hyperphosphorylated Rb is completely dissociated from E2F, enabling further expression of a wide range of E2F target genes are required for driving cells to proceed into S phase [1]. Recently, it has been identified that cyclin D-Cdk4/6 binds to a C-terminal alpha-helix region of Rb that is only distinguishable to cyclin D rather than other cyclins, cyclin E, A and B.[26] This observation based on the structural analysis of Rb phosphorylation supports that Rb is phosphorylated in a different level through multiple Cyclin-Cdk complexes. This also makes feasible the current model of a simultaneous switch-like inactivation of all mono-phosphorylated Rb isoforms through one type of Rb hyper-phosphorylation mechanism. In addition, mutational analysis of the cyclin D- Cdk 4/6 specific Rb C-terminal helix shows that disruptions of cyclin D-Cdk 4/6 binding to Rb prevents Rb phosphorylation, arrests cells in G1, and bolsters Rb's functions in tumor suppressor.[26] This cyclin-Cdk driven cell cycle transitional mechanism governs a cell committed to the cell cycle that allows cell proliferation. A cancerous cell growth often accompanies with deregulation of Cyclin D-Cdk 4/6 activity.
The hyperphosphorylated Rb dissociates from the E2F/DP1/Rb complex (which was bound to the E2F responsive genes, effectively "blocking" them from transcription), activating E2F. Activation of E2F results in transcription of various genes like cyclin E, cyclin A, DNA polymerase, thymidine kinase, etc. Cyclin E thus produced binds to CDK2, forming the cyclin E-CDK2 complex, which pushes the cell from G1 to S phase (G1/S, which initiates the G2/M transition).[27] Cyclin B-cdk1 complex activation causes breakdown of nuclear envelope and initiation of prophase, and subsequently, its deactivation causes the cell to exit mitosis.[14] A quantitative study of E2F transcriptional dynamics at the single-cell level by using engineered fluorescent reporter cells provided a quantitative framework for understanding the control logic of cell cycle entry, challenging the canonical textbook model. Genes that regulate the amplitude of E2F accumulation, such as Myc, determine the commitment in cell cycle and S phase entry. G1 cyclin-CDK activities are not the driver of cell cycle entry. Instead, they primarily tune the timing of E2F increase, thereby modulating the pace of cell cycle progression.[15]
Inhibitors
Endogenous
Two families of genes, the cip/kip (CDK interacting protein/Kinase inhibitory protein) family and the INK4a/ARF (Inhibitor of Kinase 4/Alternative Reading Frame) family, prevent the progression of the cell cycle. Because these genes are instrumental in prevention of tumor formation, they are known as tumor suppressors.
The cip/kip family includes the genes p21, p27 and p57. They halt the cell cycle in G1 phase by binding to and inactivating cyclin-CDK complexes. p21 is activated by p53 (which, in turn, is triggered by DNA damage e.g. due to radiation). p27 is activated by Transforming Growth Factor β (TGF β), a growth inhibitor.
The INK4a/ARF family includes p16INK4a, which binds to CDK4 and arrests the cell cycle in G1 phase, and p14ARF which prevents p53 degradation.
Synthetic
Synthetic inhibitors of Cdc25 could also be useful for the arrest of cell cycle and therefore be useful as antineoplastic and anticancer agents.[28]
In often cases, many human cancers possess the hyper-activated Cdk 4/6 activities.[29] Given the observations of cyclin D-Cdk 4/6 functions, inhibition of Cdk 4/6 should result in preventing a malignant tumor from proliferating. Consequently, scientists have tried to invent the synthetic Cdk4/6 inhibitor as Cdk4/6 has been characterized to be a therapeutic target for anti-tumor effectiveness. Three Cdk4/6 inhibitors - palbociclib, ribociclib, and abemaciclib - currently received FDA approval for clinical use to treat advanced-stage or metastatic, hormone-receptor-positive (HR-positive, HR+), HER2-negative (HER2-) breast cancer.[30][31] For example, palbociclib is an orally active CDK4/6 inhibitor which has demonstrated improved outcomes for ER-positive/HER2-negative advanced breast cancer. The main side effect is neutropenia which can be managed by dose reduction.[32]
Cdk4/6 targeted therapy will only treat cancer types where Rb is expressed. Cancer cells with loss of Rb have primary resistance to Cdk4/6 inhibitors.
Transcriptional regulatory network
Current evidence suggests that a semi-autonomous transcriptional network acts in concert with the CDK-cyclin machinery to regulate the cell cycle. Several gene expression studies in Saccharomyces cerevisiae have identified 800–1200 genes that change expression over the course of the cell cycle.[13][33][34] They are transcribed at high levels at specific points in the cell cycle, and remain at lower levels throughout the rest of the cycle. While the set of identified genes differs between studies due to the computational methods and criteria used to identify them, each study indicates that a large portion of yeast genes are temporally regulated.[35]
Many periodically expressed genes are driven by transcription factors that are also periodically expressed. One screen of single-gene knockouts identified 48 transcription factors (about 20% of all non-essential transcription factors) that show cell cycle progression defects.[36] Genome-wide studies using high throughput technologies have identified the transcription factors that bind to the promoters of yeast genes, and correlating these findings with temporal expression patterns have allowed the identification of transcription factors that drive phase-specific gene expression.[33][37] The expression profiles of these transcription factors are driven by the transcription factors that peak in the prior phase, and computational models have shown that a CDK-autonomous network of these transcription factors is sufficient to produce steady-state oscillations in gene expression).[34][38]
Experimental evidence also suggests that gene expression can oscillate with the period seen in dividing wild-type cells independently of the CDK machinery. Orlando et al. used microarrays to measure the expression of a set of 1,271 genes that they identified as periodic in both wild type cells and cells lacking all S-phase and mitotic cyclins (clb1,2,3,4,5,6). Of the 1,271 genes assayed, 882 continued to be expressed in the cyclin-deficient cells at the same time as in the wild type cells, despite the fact that the cyclin-deficient cells arrest at the border between G1 and S phase. However, 833 of the genes assayed changed behavior between the wild type and mutant cells, indicating that these genes are likely directly or indirectly regulated by the CDK-cyclin machinery. Some genes that continued to be expressed on time in the mutant cells were also expressed at different levels in the mutant and wild type cells. These findings suggest that while the transcriptional network may oscillate independently of the CDK-cyclin oscillator, they are coupled in a manner that requires both to ensure the proper timing of cell cycle events.[34] Other work indicates that phosphorylation, a post-translational modification, of cell cycle transcription factors by Cdk1 may alter the localization or activity of the transcription factors in order to tightly control timing of target genes.[36][39][40]
While oscillatory transcription plays a key role in the progression of the yeast cell cycle, the CDK-cyclin machinery operates independently in the early embryonic cell cycle. Before the midblastula transition, zygotic transcription does not occur and all needed proteins, such as the B-type cyclins, are translated from maternally loaded mRNA.[41]
DNA replication and DNA replication origin activity
Analyses of synchronized cultures of Saccharomyces cerevisiae under conditions that prevent DNA replication initiation without delaying cell cycle progression showed that origin licensing decreases the expression of genes with origins near their 3' ends, revealing that downstream origins can regulate the expression of upstream genes.[42] This confirms previous predictions from mathematical modeling of a global causal coordination between DNA replication origin activity and mRNA expression,[43][44][45] and shows that mathematical modeling of DNA microarray data can be used to correctly predict previously unknown biological modes of regulation.
Checkpoints
Cell cycle checkpoints are used by the cell to monitor and regulate the progress of the cell cycle.[46] Checkpoints prevent cell cycle progression at specific points, allowing verification of necessary phase processes and repair of DNA damage. The cell cannot proceed to the next phase until checkpoint requirements have been met. Checkpoints typically consist of a network of regulatory proteins that monitor and dictate the progression of the cell through the different stages of the cell cycle.
There are several checkpoints to ensure that damaged or incomplete DNA is not passed on to daughter cells. Three main checkpoints exist: the G1/S checkpoint, the G2/M checkpoint and the metaphase (mitotic) checkpoint.
G1/S transition is a rate-limiting step in the cell cycle and is also known as restriction point.[14] This is where the cell checks whether it has enough raw materials to fully replicate its DNA (nucleotide bases, DNA synthase, chromatin, etc.). An unhealthy or malnourished cell will get stuck at this checkpoint.
The G2/M checkpoint is where the cell ensures that it has enough cytoplasm and phospholipids for two daughter cells. But sometimes more importantly, it checks to see if it is the right time to replicate. There are some situations where many cells need to all replicate simultaneously (for example, a growing embryo should have a symmetric cell distribution until it reaches the mid-blastula transition). This is done by controlling the G2/M checkpoint.
The metaphase checkpoint is a fairly minor checkpoint, in that once a cell is in metaphase, it has committed to undergoing mitosis. However that's not to say it isn't important. In this checkpoint, the cell checks to ensure that the spindle has formed and that all of the chromosomes are aligned at the spindle equator before anaphase begins.[47]
While these are the three "main" checkpoints, not all cells have to pass through each of these checkpoints in this order to replicate. Many types of cancer are caused by mutations that allow the cells to speed through the various checkpoints or even skip them altogether. Going from S to M to S phase almost consecutively. Because these cells have lost their checkpoints, any DNA mutations that may have occurred are disregarded and passed on to the daughter cells. This is one reason why cancer cells have a tendency to exponentially accrue mutations. Aside from cancer cells, many fully differentiated cell types no longer replicate so they leave the cell cycle and stay in G0 until their death. Thus removing the need for cellular checkpoints. An alternative model of the cell cycle response to DNA damage has also been proposed, known as the postreplication checkpoint.
Checkpoint regulation plays an important role in an organism's development. In sexual reproduction, when egg fertilization occurs, when the sperm binds to the egg, it releases signalling factors that notify the egg that it has been fertilized. Among other things, this induces the now fertilized oocyte to return from its previously dormant, G0, state back into the cell cycle and on to mitotic replication and division.
p53 plays an important role in triggering the control mechanisms at both G1/S and G2/M checkpoints. In addition to p53, checkpoint regulators are being heavily researched for their roles in cancer growth and proliferation.
Fluorescence imaging of the cell cycle
.gif)
Pioneering work by Atsushi Miyawaki and coworkers developed the fluorescent ubiquitination-based cell cycle indicator (FUCCI), which enables fluorescence imaging of the cell cycle. Originally, a green fluorescent protein, mAG, was fused to hGem(1/110) and an orange fluorescent protein (mKO2) was fused to hCdt1(30/120). Note, these fusions are fragments that contain a nuclear localization signal and ubiquitination sites for degradation, but are not functional proteins. The green fluorescent protein is made during the S, G2, or M phase and degraded during the G0 or G1 phase, while the orange fluorescent protein is made during the G0 or G1 phase and destroyed during the S, G2, or M phase.[48] A far-red and near-infrared FUCCI was developed using a cyanobacteria-derived fluorescent protein (smURFP) and a bacteriophytochrome-derived fluorescent protein (movie found at this link).[49]
Role in tumor formation
A disregulation of the cell cycle components may lead to tumor formation.[50] As mentioned above, when some genes like the cell cycle inhibitors, RB, p53 etc. mutate, they may cause the cell to multiply uncontrollably, forming a tumor. Although the duration of cell cycle in tumor cells is equal to or longer than that of normal cell cycle, the proportion of cells that are in active cell division (versus quiescent cells in G0 phase) in tumors is much higher than that in normal tissue. Thus there is a net increase in cell number as the number of cells that die by apoptosis or senescence remains the same.
The cells which are actively undergoing cell cycle are targeted in cancer therapy as the DNA is relatively exposed during cell division and hence susceptible to damage by drugs or radiation. This fact is made use of in cancer treatment; by a process known as debulking, a significant mass of the tumor is removed which pushes a significant number of the remaining tumor cells from G0 to G1 phase (due to increased availability of nutrients, oxygen, growth factors etc.). Radiation or chemotherapy following the debulking procedure kills these cells which have newly entered the cell cycle.[14]
The fastest cycling mammalian cells in culture, crypt cells in the intestinal epithelium, have a cycle time as short as 9 to 10 hours. Stem cells in resting mouse skin may have a cycle time of more than 200 hours. Most of this difference is due to the varying length of G1, the most variable phase of the cycle. M and S do not vary much.
In general, cells are most radiosensitive in late M and G2 phases and most resistant in late S phase.
For cells with a longer cell cycle time and a significantly long G1 phase, there is a second peak of resistance late in G1.
The pattern of resistance and sensitivity correlates with the level of sulfhydryl compounds in the cell. Sulfhydryls are natural substances that protect cells from radiation damage and tend to be at their highest levels in S and at their lowest near mitosis.
Homologous recombination (HR) is an accurate process for repairing DNA double-strand breaks. HR is nearly absent in G1 phase, is most active in S phase, and declines in G2/M.[51] Non-homologous end joining, a less accurate and more mutagenic process for repairing double strand breaks, is active throughout the cell cycle.
See also
- Cellular model
- Eukaryotic DNA replication
- Origin recognition complex
- Retinoblastoma protein
- Synchronous culture – synchronization of cell cultures
- Wee1
References
- Wang JD, Levin PA (November 2009). "Metabolism, cell growth and the bacterial cell cycle". Nature Reviews. Microbiology. 7 (11): 822–7. doi:10.1038/nrmicro2202. PMC 2887316. PMID 19806155.
- Cooper GM (2000). "Chapter 14: The Eukaryotic Cell Cycle". The cell: a molecular approach (2nd ed.). Washington, D.C: ASM Press. ISBN 978-0-87893-106-4.
- Smith JA, Martin L (April 1973). "Do cells cycle?". Proceedings of the National Academy of Sciences of the United States of America. 70 (4): 1263–7. Bibcode:1973PNAS...70.1263S. doi:10.1073/pnas.70.4.1263. PMC 433472. PMID 4515625.
- Wu RS, Bonner WM (December 1981). "Separation of basal histone synthesis from S-phase histone synthesis in dividing cells". Cell. 27 (2 Pt 1): 321–30. doi:10.1016/0092-8674(81)90415-3. PMID 7199388.
- Nelson DM, Ye X, Hall C, Santos H, Ma T, Kao GD, et al. (November 2002). "Coupling of DNA synthesis and histone synthesis in S phase independent of cyclin/cdk2 activity". Molecular and Cellular Biology. 22 (21): 7459–72. doi:10.1128/MCB.22.21.7459-7472.2002. PMC 135676. PMID 12370293.
- Cameron IL, Greulich RC (July 1963). "Evidence for an essentially constant duration of DNA synthesis in renewing epithelia of the adult mouse". The Journal of Cell Biology. 18: 31–40. doi:10.1083/jcb.18.1.31. PMC 2106275. PMID 14018040.
- Rubenstein, Irwin, and Susan M. Wick. "Cell." World Book Online Reference Center. 2008. 12 January 2008 <"Archived copy". Archived from the original on 30 May 2011. Retrieved 2009-07-10.CS1 maint: archived copy as title (link)>
- Maton A, Lahart D, Hopkins J, Warner MQ, Johnson S, Wright JD (1997). Cells: Building Blocks of Life. New Jersey: Prentice Hall. pp. 70–4. ISBN 978-0-13-423476-2.
- De Souza CP, Osmani SA (September 2007). "Mitosis, not just open or closed". Eukaryotic Cell. 6 (9): 1521–7. doi:10.1128/EC.00178-07. PMC 2043359. PMID 17660363.
- Lilly MA, Duronio RJ (April 2005). "New insights into cell cycle control from the Drosophila endocycle". Oncogene. 24 (17): 2765–75. doi:10.1038/sj.onc.1208610. PMID 15838513.
- Nigg EA (June 1995). "Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle". BioEssays. 17 (6): 471–80. doi:10.1002/bies.950170603. PMID 7575488.
- "Press release". Nobelprize.org.
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, et al. (December 1998). "Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization". Molecular Biology of the Cell. 9 (12): 3273–97. doi:10.1091/mbc.9.12.3273. PMC 25624. PMID 9843569.
- Robbins SL, Cotran RS (2004). Kumar V, Abbas AK, Fausto N (eds.). Pathological Basis of Disease. Elsevier. ISBN 978-81-8147-528-2.
- Dong P, Maddali MV, Srimani JK, Thélot F, Nevins JR, Mathey-Prevot B, You L (September 2014). "Division of labour between Myc and G1 cyclins in cell cycle commitment and pace control". Nature Communications. 5: 4750. Bibcode:2014NatCo...5.4750D. doi:10.1038/ncomms5750. PMC 4164785. PMID 25175461.
- Mahmoudi M, Azadmanesh K, Shokrgozar MA, Journeay WS, Laurent S (May 2011). "Effect of nanoparticles on the cell life cycle". Chemical Reviews. 111 (5): 3407–32. doi:10.1021/cr1003166. PMID 21401073.
- Goel S, DeCristo MJ, McAllister SS, Zhao JJ (November 2018). "CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest". Trends in Cell Biology. 28 (11): 911–925. doi:10.1016/j.tcb.2018.07.002. PMC 6689321. PMID 30061045.
- Burkhart DL, Sage J (September 2008). "Cellular mechanisms of tumour suppression by the retinoblastoma gene". Nature Reviews. Cancer. 8 (9): 671–82. doi:10.1038/nrc2399. PMC 6996492. PMID 18650841.
- Morgan DO (2007). The cell cycle : principles of control. London: New Science Press. ISBN 978-0-19-920610-0. OCLC 70173205.
- Paternot S, Bockstaele L, Bisteau X, Kooken H, Coulonval K, Roger PP (February 2010). "Rb inactivation in cell cycle and cancer: the puzzle of highly regulated activating phosphorylation of CDK4 versus constitutively active CDK-activating kinase". Cell Cycle. 9 (4): 689–99. doi:10.4161/cc.9.4.10611. PMID 20107323.
- Henley SA, Dick FA (March 2012). "The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle". Cell Division. 7 (1): 10. doi:10.1186/1747-1028-7-10. PMC 3325851. PMID 22417103.
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF (June 2014). "Cyclin D activates the Rb tumor suppressor by mono-phosphorylation". eLife. 3: e02872. doi:10.7554/eLife.02872. PMC 4076869. PMID 24876129.
- Morris EJ, Dyson NJ (1 January 2001). Retinoblastoma protein partners. Advances in Cancer Research. 82. Academic Press. pp. 1–54. doi:10.1016/s0065-230x(01)82001-7. ISBN 9780120066827. PMID 11447760.
- Dyson NJ (July 2016). "RB1: a prototype tumor suppressor and an enigma". Genes & Development. 30 (13): 1492–502. doi:10.1101/gad.282145.116. PMC 4949322. PMID 27401552.
- Sanidas I, Morris R, Fella KA, Rumde PH, Boukhali M, Tai EC, et al. (March 2019). "A Code of Mono-phosphorylation Modulates the Function of RB". Molecular Cell. 73 (5): 985–1000.e6. doi:10.1016/j.molcel.2019.01.004. PMC 6424368. PMID 30711375.
- Topacio BR, Zatulovskiy E, Cristea S, Xie S, Tambo CS, Rubin SM, et al. (May 2019). "Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein's C-Terminal Helix". Molecular Cell. 74 (4): 758–770.e4. doi:10.1016/j.molcel.2019.03.020. PMC 6800134. PMID 30982746.
- Norbury C (1995). "Cdk2 protein kinase (vertebrates)". In Hardie DG, Hanks S (eds.). Protein kinase factsBook. Boston: Academic Press. pp. 184. ISBN 978-0-12-324719-3.
- "Presentation on CDC25 PHOSPHATASES: A Potential Target for Novel Anticancer Agents". Archived from the original on 3 March 2016. Retrieved 11 March 2010.
- Sherr CJ, Beach D, Shapiro GI (April 2016). "Targeting CDK4 and CDK6: From Discovery to Therapy". Cancer Discovery. 6 (4): 353–67. doi:10.1158/2159-8290.cd-15-0894. PMC 4821753. PMID 26658964.
- O'Leary B, Finn RS, Turner NC (July 2016). "Treating cancer with selective CDK4/6 inhibitors". Nature Reviews. Clinical Oncology. 13 (7): 417–30. doi:10.1038/nrclinonc.2016.26. PMID 27030077.
- Bilgin B, Sendur MA, Şener Dede D, Akıncı MB, Yalçın B (September 2017). "A current and comprehensive review of cyclin-dependent kinase inhibitors for the treatment of metastatic breast cancer". Current Medical Research and Opinion. 33 (9): 1559–1569. doi:10.1080/03007995.2017.1348344. PMID 28657360.
- Schmidt M, Sebastian M (August 2018). "Palbociclib-The First of a New Class of Cell Cycle Inhibitors". Recent Results in Cancer Research. Fortschritte der Krebsforschung. Progres dans les Recherches Sur le Cancer. Recent Results in Cancer Research. 211: 153–175. doi:10.1007/978-3-319-91442-8_11. ISBN 978-3-319-91441-1. PMID 30069766.
- Pramila T, Wu W, Miles S, Noble WS, Breeden LL (August 2006). "The Forkhead transcription factor Hcm1 regulates chromosome segregation genes and fills the S-phase gap in the transcriptional circuitry of the cell cycle". Genes & Development. 20 (16): 2266–78. doi:10.1101/gad.1450606. PMC 1553209. PMID 16912276.
- Orlando DA, Lin CY, Bernard A, Wang JY, Socolar JE, Iversen ES, et al. (June 2008). "Global control of cell-cycle transcription by coupled CDK and network oscillators". Nature. 453 (7197): 944–7. Bibcode:2008Natur.453..944O. doi:10.1038/nature06955. PMC 2736871. PMID 18463633.
- de Lichtenberg U, Jensen LJ, Fausbøll A, Jensen TS, Bork P, Brunak S (April 2005). "Comparison of computational methods for the identification of cell cycle-regulated genes". Bioinformatics. 21 (7): 1164–71. doi:10.1093/bioinformatics/bti093. PMID 15513999.
- White MA, Riles L, Cohen BA (February 2009). "A systematic screen for transcriptional regulators of the yeast cell cycle". Genetics. 181 (2): 435–46. doi:10.1534/genetics.108.098145. PMC 2644938. PMID 19033152.
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, et al. (October 2002). "Transcriptional regulatory networks in Saccharomyces cerevisiae". Science. 298 (5594): 799–804. doi:10.1126/science.1075090. PMID 12399584.
- Simon I, Barnett J, Hannett N, Harbison CT, Rinaldi NJ, Volkert TL, et al. (September 2001). "Serial regulation of transcriptional regulators in the yeast cell cycle". Cell. 106 (6): 697–708. doi:10.1016/S0092-8674(01)00494-9. PMID 11572776.
- Sidorova JM, Mikesell GE, Breeden LL (December 1995). "Cell cycle-regulated phosphorylation of Swi6 controls its nuclear localization". Molecular Biology of the Cell. 6 (12): 1641–58. doi:10.1091/mbc.6.12.1641. PMC 301322. PMID 8590795.
- Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, et al. (October 2003). "Targets of the cyclin-dependent kinase Cdk1". Nature. 425 (6960): 859–64. Bibcode:2003Natur.425..859U. doi:10.1038/nature02062. PMID 14574415.
- Morgan DO (2007). "2–3". The Cell Cycle: Principles of Control. London: New Science Press. p. 18. ISBN 978-0-9539181-2-6.
- Omberg L, Meyerson JR, Kobayashi K, Drury LS, Diffley JF, Alter O (October 2009). "Global effects of DNA replication and DNA replication origin activity on eukaryotic gene expression". Molecular Systems Biology. 5: 312. doi:10.1038/msb.2009.70. PMC 2779084. PMID 19888207.
- Alter O, Golub GH, Brown PO, Botstein D (2004). Deutscher MP, Black S, Boehmer PE, D'Urso G, Fletcher TM, Huijing F, Marshall A, Pulverer B, Renault B, Rosenblatt JD, Slingerland JM, Whelan WJ (eds.). "Novel Genome-Scale Correlation between DNA Replication and RNA Transcription During the Cell Cycle in Yeast is Predicted by Data-Driven Models" (PDF). Miami Nature Biotechnology Winter Symposium: Cell Cycle, Chromosomes and Cancer. Miami Beach, FL: University of Miami School of Medicine, vol. 15 (January 31 – February 4, 2004). Cite journal requires
|journal=(help) - Alter O, Golub GH (November 2004). "Integrative analysis of genome-scale data by using pseudoinverse projection predicts novel correlation between DNA replication and RNA transcription". Proceedings of the National Academy of Sciences of the United States of America. 101 (47): 16577–82. Bibcode:2004PNAS..10116577A. doi:10.1073/pnas.0406767101. PMC 534520. PMID 15545604.
- Omberg L, Golub GH, Alter O (November 2007). "A tensor higher-order singular value decomposition for integrative analysis of DNA microarray data from different studies". Proceedings of the National Academy of Sciences of the United States of America. 104 (47): 18371–6. Bibcode:2007PNAS..10418371O. doi:10.1073/pnas.0709146104. PMC 2147680. PMID 18003902.
- Elledge SJ (December 1996). "Cell cycle checkpoints: preventing an identity crisis". Science. 274 (5293): 1664–72. Bibcode:1996Sci...274.1664E. doi:10.1126/science.274.5293.1664. PMID 8939848.
- LeMaire-Adkins R, Radke K, Hunt PA (December 1997). "Lack of checkpoint control at the metaphase/anaphase transition: a mechanism of meiotic nondisjunction in mammalian females". The Journal of Cell Biology. 139 (7): 1611–9. doi:10.1083/jcb.139.7.1611. PMC 2132649. PMID 9412457.
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, et al. (February 2008). "Visualizing spatiotemporal dynamics of multicellular cell-cycle progression". Cell. 132 (3): 487–98. doi:10.1016/j.cell.2007.12.033. PMID 18267078.
- Rodriguez EA, Tran GN, Gross LA, Crisp JL, Shu X, Lin JY, Tsien RY (September 2016). "A far-red fluorescent protein evolved from a cyanobacterial phycobiliprotein". Nature Methods. 13 (9): 763–9. doi:10.1038/nmeth.3935. PMC 5007177. PMID 27479328.
- Champeris Tsaniras S, Kanellakis N, Symeonidou IE, Nikolopoulou P, Lygerou Z, Taraviras S (June 2014). "Licensing of DNA replication, cancer, pluripotency and differentiation: an interlinked world?". Seminars in Cell & Developmental Biology. 30: 174–80. doi:10.1016/j.semcdb.2014.03.013. PMID 24641889.
- Mao Z, Bozzella M, Seluanov A, Gorbunova V (September 2008). "DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells". Cell Cycle. 7 (18): 2902–6. doi:10.4161/cc.7.18.6679. PMC 2754209. PMID 18769152.
Further reading
- Morgan DO (2007). The Cell Cycle: Principles of Control. London: Published by New Science Press in association with Oxford University Press. ISBN 978-0-87893-508-6.
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2008). "Chapter 17". Molecular Biology of the Cell (5th ed.). New York: Garland Science. ISBN 978-0-8153-4111-6.
- Krieger M, Scott MP, Matsudaira PT, Lodish HF, Darnell JE, Zipursky L, Kaiser C, Berk A (2004). Molecular cell biology. New York: W.H. Freeman and CO. ISBN 978-0-7167-4366-8.
- Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R (2004). "Chapter 7". Molecular biology of the gene (5th ed.). San Francisco: Pearson/Benjamin Cummings. ISBN 978-0-8053-4642-8.
External links
| Wikimedia Commons has media related to Cell cycle. |

- David Morgan's Seminar: Controlling the Cell Cycle
- The cell cycle & Cell death
- Transcriptional program of the cell cycle: high-resolution timing
- Cell cycle and metabolic cycle regulated transcription in yeast
- Cell Cycle Animation 1Lec.com
- Cell Cycle
- Fucci:Using GFP to visualize the cell-cycle
- Science Creative Quarterly's overview of the cell cycle
- KEGG – Human Cell Cycle