Carbon-13 nuclear magnetic resonance
Carbon-13 (C13) nuclear magnetic resonance (most commonly known as carbon-13 NMR or 13C NMR or sometimes simply referred to as carbon NMR) is the application of nuclear magnetic resonance (NMR) spectroscopy to carbon. It is analogous to proton NMR (1
H
NMR) and allows the identification of carbon atoms in an organic molecule just as proton NMR identifies hydrogen atoms. As such 13C NMR is an important tool in chemical structure elucidation in organic chemistry. 13C NMR detects only the 13
C
isotope of carbon, whose natural abundance is only 1.1%, because the main carbon isotope, 12
C
, is not detectable by NMR since its nucleus has zero spin.
Chemical shifts
13C chemical shifts follow the same principles as those of 1H, although the typical range of chemical shifts is much larger than for 1H (by a factor of about 20). The chemical shift reference standard for 13C is the carbons in tetramethylsilane (TMS), [1] whose chemical shift is considered to be 0.0 ppm.
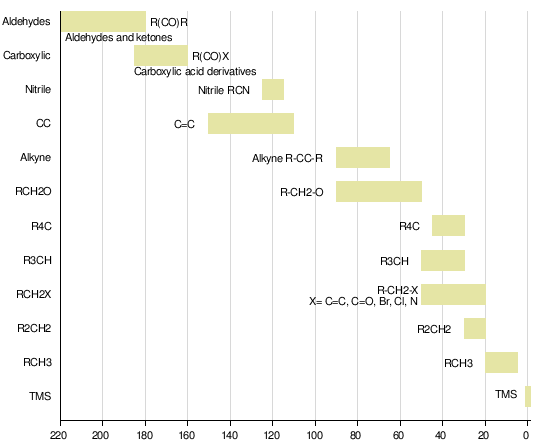
Typical chemical shifts in 13C-NMR
Implementation
Sensitivity
13C NMR has a number of complications that are not encountered in proton NMR. 13C NMR is much less sensitive to carbon than 1H NMR is to hydrogen since the major isotope of carbon, the 12C isotope, has a spin quantum number of zero and so is not magnetically active and therefore not detectable by NMR. Only the much less common 13C isotope, present naturally at 1.1% natural abundance, is magnetically active with a spin quantum number of 1/2 (like 1H) and therefore detectable by NMR. Therefore, only the few 13C nuclei present resonate in the magnetic field, although this can be overcome by isotopic enrichment of e.g. protein samples. In addition, the gyromagnetic ratio (6.728284 107 rad T−1 s−1) is only 1/4 that of 1H, further reducing the sensitivity. The overall receptivity of 13C is about 4 orders of magnitude lower than 1H.[2]
High field magnets with internal bores capable of accepting larger sample tubes (typically 10 mm in diameter for 13C NMR versus 5 mm for 1H NMR), the use of relaxation reagents,[3] for example Cr(acac)3 (Chromium(III) acetylacetonate), and appropriate pulse sequences have reduced the time needed to acquire quantitative spectra and have made quantitative carbon-13 NMR a commonly used technique in many industrial labs. Applications range from quantification of drug purity to determination of the composition of high molecular weight synthetic polymers.
In a typical run on an organic compound, a 13C NMR may require several hours to record the spectrum of a one-milligram sample, compared to 15–30 minutes for 1H NMR, and that spectrum would be of lower quality. The nuclear dipole is weaker, the difference in energy between alpha and beta states is one-quarter that of proton NMR, and the Boltzmann population difference is correspondingly less.[4]
Coupling modes
Another potential complication results from the presence of large one bond J-coupling constants between carbon and hydrogen (typically from 100 to 250 Hz). In order to suppress these couplings, which would otherwise complicate the spectra and further reduce sensitivity, carbon NMR spectra are usually proton decoupled to remove the signal splitting. Couplings between carbons can be ignored due to the low natural abundance of 13C. Hence in contrast to typical proton NMR spectra which show multiplets for each proton position, carbon NMR spectra show a single peak for each chemically non-equivalent carbon atom.[5]
In further contrast to 1H NMR, the intensities of the signals are not normally proportional to the number of equivalent 13C atoms and are instead strongly dependent on the number of surrounding spins (typically 1H). Spectra can be made more quantitative if necessary by allowing sufficient time for the nuclei to relax between repeat scans.
The most common modes of recording 13C spectra are proton-noise decoupling (also known as noise, proton, or broadband decoupling), off-resonance decoupling, and gated decoupling. These modes are meant to address the large J values for 13C - H (110–320 Hz), 13C - C - H (5–60 Hz), and 13C - C - C - H (5–25 Hz) which otherwise make completely proton coupled 13C spectra difficult to interpret.[6]
With proton-noise decoupling, in which most spectra are run, a noise decoupler strongly irradiates the sample with a broad (approximately 1000 Hz) range of radio frequencies covering the range (such as 100 MHz for a 23,486 gauss field) at which protons change their nuclear spin. The rapid changes in proton spin create an effective heteronuclear decoupling, increasing carbon signal strength on account of the nuclear Overhauser effect (NOE) and simplifying the spectrum so that each nonequivalent carbon produces a singlet peak. The relative intensities are unreliable because some carbons have a larger spin-lattice relaxation time and others have weaker NOE enhancement.[6]
In gated decoupling, the noise decoupler is gated on early in the free induction delay but gated off for the pulse delay. This largely prevents NOE enhancement, allowing the strength of individual 13C peaks to be meaningfully compared by integration, at a cost of half to two-thirds of the overall sensitivity.[6]
With off-resonance decoupling, the noise decoupler irradiates the sample at 1000–2000 Hz upfield or 2000–3000 Hz downfield of the proton resonance frequency. This retains couplings between protons immediately adjacent to 13C atoms but most often removes the others, allowing narrow multiplets to be visualized with one extra peak per bound proton (unless bound methylene protons are nonequivalent, in which case a pair of doublets may be observed).[6]
Distortionless enhancement by polarization transfer spectra
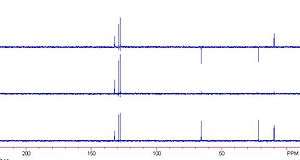
From top to bottom: 135°, 90° and 45°
Distortionless enhancement by polarization transfer (DEPT)[7] is an NMR method used for determining the presence of primary, secondary and tertiary carbon atoms. The DEPT experiment differentiates between CH, CH2 and CH3 groups by variation of the selection angle parameter (the tip angle of the final 1H pulse): 135° angle gives all CH and CH3 in a phase opposite to CH2; 90° angle gives only CH groups, the others being suppressed; 45° angle gives all carbons with attached protons (regardless of number) in phase.
Signals from quaternary carbons and other carbons with no attached protons are always absent (due to the lack of attached protons).
The polarization transfer from 1H to 13C has the secondary advantage of increasing the sensitivity over the normal 13C spectrum (which has a modest enhancement from the nuclear overhauser effect (NOE) due to the 1H decoupling).
Attached proton test spectra
Another useful way of determining how many protons a carbon in a molecule is bonded to is to use an attached proton test (APT), which distinguishes between carbon atoms with even or odd number of attached hydrogens. A proper spin-echo sequence is able to distinguish between S, I2S and I1S, I3S spin systems: the first will appear as positive peaks in the spectrum, while the latter as negative peaks (pointing downwards), while retaining relative simplicity in the spectrum since it is still broadband proton decoupled.
Even though this technique does not distinguish fully between CHn groups, it is so easy and reliable that it is frequently employed as a first attempt to assign peaks in the spectrum and elucidate the structure.[8] Additionally, signals from quaternary carbons and other carbons with no attached protons are still detectable, so in many cases an additional conventional 13C spectrum is not required, which is an advantage over DEPT. It is, however, sometimes possible that a CH and CH2 signal have coincidentally equivalent chemical shifts resulting in annulment in the APT spectrum due to the opposite phases. For this reason the conventional 13C{1H} spectrum or HSQC are occasionally also acquired.
See also
References
- The Theory of NMR - Chemical Shift
- R. M. Silverstein; G. C. Bassler; T. C. Morrill (1991). Spectrometric Identification of Organic Compounds. Wiley.
- Caytan E, Remaud GS, Tenailleau E, Akoka S (2007). "Precise and accurate quantitative 13C NMR with reduced experimental time". Talanta. 71 (3): 1016–1021. doi:10.1016/j.talanta.2006.05.075. PMID 19071407.
- "Measuring 13C NMR Spectra". University of Wisconsin.
- "Introduction to Carbon NMR". University of Puget Sound.
- Lal Dhar Singh Yadav (2013-08-13). Organic Spectroscopy. Springer. pp. 197–199. ISBN 9781402025754.
- Doddrell, D.M.; Pegg, D.T.; Bendall, M.R. (1982). "Distortionless enhancement of NMR signals by polarization transfer". J. Magn. Reson. 48 (2): 323–327. Bibcode:1982JMagR..48..323D. doi:10.1016/0022-2364(82)90286-4.
- Keeler, James (2010). Understanding NMR Spectroscopy (2nd ed.). John Wiley & Sons. p. 457. ISBN 978-0-470-74608-0.
External links
- Carbon NMR spectra, where there are three spectra of ethyl phthalate, ethyl ester of orthophthalic acid: completely coupled, completely decoupled and off-resonance decoupled (in this order).
- For an extended tabulation of 13C shifts and coupling constants.