J-coupling
In nuclear chemistry and nuclear physics, Scalar or J-couplings (also called indirect dipole–dipole coupling) are mediated through chemical bonds connecting two spins. It is an indirect interaction between two nuclear spins which arises from hyperfine interactions between the nuclei and local electrons.[1] In NMR spectroscopy J-coupling contains information about relative bond distances and angles. Most importantly, J-coupling provides information on the connectivity of chemical bonds. It is responsible for the often complex splitting of resonance lines in the NMR spectra of fairly simple molecules.
Vector model and manifestations for chemical structure assignments
The origin of J-coupling can be visualized by a vector model for a simple molecule such as hydrogen fluoride (HF). In HF, the two nuclei have spin 1/2. Four states are possible, depending on the relative alignment of the H and F nuclear spins with the external magnetic field. The selection rules of NMR spectroscopy dictate that ΔI = 1, which means that a given photon (in the radio frequency range) can affect ("flip") only one of the two nuclear spins.
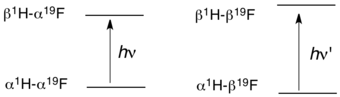
J-coupling provides three parameters: the multiplicity (the "number of lines"), the magnitude of the coupling (strong, medium, weak), and the sign of the coupling.
Multiplicity
The multiplicity provides information on the number of centers coupled to the signal of interest, and their nuclear spin. For simple systems, as in 1H-1H coupling in NMR spectroscopy, the multiplicity reflects the number of adjacent, magnetically nonequivalent protons. Nuclei with spins greater than 1/2, which are called quadrupolar, can give rise to greater splitting, although in many cases coupling to quadrupolar nuclei is not observed. Many elements consist of nuclei with nuclear spin and without. In these cases the observed spectrum is the sum of spectra for each isotopomer. One of the great conveniences of NMR spectroscopy for organic molecules is that several important lighter spin 1/2 nuclei are either monoisotopic, e.g. 31P and 19F, or have very high natural abundance, e.g. 1H. An additional convenience is that 12C and 16O have no nuclear spin so these nuclei, which are common in organic molecules, do not cause splitting patterns in NMR.
Magnitude of J-coupling
For 1H–1H coupling, the magnitude of J provides information on the proximity of the coupling partners. Generally speaking 2-bond coupling (i.e. 1H–C–1H) is stronger than three-bond coupling (1H–C–C–1H). The magnitude of the coupling also provides information on the dihedral angles relating the coupling partners, as described by the Karplus relationship.
For heteronuclear coupling, the magnitude of J is related to the nuclear magnetic moments of the coupling partners. 19F, with a high nuclear magnetic moment, gives rise to large coupling to protons. 103Rh, with a very small nuclear magnetic moment, gives only small couplings to 1H. To correct for the effect of the nuclear magnetic moment (or equivalently the gyromagnetic ratio γ), the "reduced coupling constant" K is often discussed, where
- K = 4π2J/hγxγy.
The value of J also has a sign, and couplings constants of comparable magnitude often have opposite signs.[2]
J-coupling Hamiltonian
The Hamiltonian of a molecular system may be taken as:
- H = D1 + D2 + D3,
- D1 = electron orbital–orbital, spin–orbital, spin–spin and electron-spin–external-field interactions
- D2 = magnetic interactions between nuclear spin and electron spin
- D3 = direct interaction of nuclei with each other
For a singlet molecular state and frequent molecular collisions, D1 and D3 are almost zero. The full form of the J-coupling interaction between spins 'Ij and Ik on the same molecule is:
- H = 2π Ij · Jjk · Ik
where Jjk is the J-coupling tensor, a real 3 × 3 matrix. It depends on molecular orientation, but in an isotropic liquid it reduces to a number, the so-called scalar coupling. In 1D NMR, the scalar coupling leads to oscillations in the free induction decay as well as splittings of lines in the spectrum.
Decoupling
By selective radio frequency irradiation, NMR spectra can be fully or partially decoupled, eliminating or selectively reducing the coupling effect. Carbon-13 NMR spectra are often recorded with proton decoupling.
History
In September 1951, H. S. Gutowsky, D. W. McCall, and C. P. Slichter reported experiments on , , and , where they explained the presence of multiple resonance lines with an interaction of the form .[3]




Independently, in October 1951, E. L. Hahn and D. E. Maxwell reported a spin echo experiment which indicates the existence of an interaction between two protons in dichloroacetaldehyde. In the echo experiment, two short, intense pulses of radiofrequency magnetic field are applied to the spin ensemble at the nuclear resonance condition and are separated by a time interval of τ. The echo appears with a given amplitude at time 2τ. For each setting of τ, the maximum value of the echo signal is measured and plotted as a function of τ. If the spin ensemble consists of a magnetic moment, a monotonic decay in the echo envelope is obtained. In the Hahn-Maxwell experiment, the decay was modulated by two frequencies: one frequency corresponded with the difference in chemical shift between the two non-equivalent spins and a second frequency, J, that was smaller and independent of magnetic field strength (J/2π = 0.7 Hz).[4] Such interaction came as a great surprise. The direct interaction between two magnetic dipoles depends on the relative position of two nuclei in such a way that when averaged over all possible orientations of the molecule it equals to zero.
In November 1951, N. F. Ramsey and E. M. Purcell proposed a mechanism that explained the observation and gave rise to an interaction of the form I1·I2. The mechanism is the magnetic interaction between each nucleus and the electron spin of its own atom together with the exchange coupling of the electron spins with each other.[5]
In the 1990s, direct evidence was found for the presence of J-couplings between magnetically active nuclei on both sides of the hydrogen bond.[6][7] Initially, it was surprising to observe such couplings across hydrogen bonds since J-couplings are usually associated with the presence of purely covalent bonds. However, it is now well established that the H-bond J-couplings follow the same electron-mediated polarization mechanism as their covalent counterparts.[8]
The spin–spin coupling between nonbonded atoms in close proximity has sometimes been observed between fluorine, nitrogen, carbon, silicon and phosphorus atoms.[9][10][11]
See also
- Earth's field NMR
- Exclusive correlation spectroscopy (ECOSY)
- Magnetic dipole–dipole interaction (dipolar coupling)
- Nuclear magnetic resonance
- Nuclear magnetic resonance spectroscopy of carbohydrates
- Nuclear magnetic resonance spectroscopy of nucleic acids
- Nuclear magnetic resonance spectroscopy of proteins
- Proton NMR
- Relaxation (NMR)
- Residual dipolar coupling
References
- Hahn, E. L.; Maxwell, D. E. (1952). "Spin Echo Measurements of Nuclear Spin Coupling in Molecules". Phys. Rev. 88 (5): 1070–1084. Bibcode:1952PhRv...88.1070H. doi:10.1103/PhysRev.88.1070.
- Pregosin, P. S.; Rueegger, H. (2004). "Nuclear magnetic resonance spectroscopy". In McCleverty, Jon A.; Thomas J., Meyer (eds.). Comprehensive Coordination Chemistry II. 2. pp. 1–35. doi:10.1016/B0-08-043748-6/01061-6.
- Gutowsky, H. S.; McCall, D. W.; Slichter, C. P. (1951). "Coupling among Nuclear Magnetic Dipoles in Molecules". Phys. Rev. 84 (3): 589–90. Bibcode:1951PhRv...84..589G. doi:10.1103/PhysRev.84.589.2.
- Hahn, E. L.; Maxwell, D. E. (1951). "Chemical Shift and Field Independent Frequency Modulation of the Spin Echo Envelope". Phys. Rev. 84 (6): 1246–1247. Bibcode:1951PhRv...84.1246H. doi:10.1103/PhysRev.84.1246.
- Ramsey, N. F.; Purcell, E. M. (1952). "Interactions between Nuclear Spins in Molecules". Phys. Rev. 85 (1): 143–144. Bibcode:1952PhRv...85..143R. doi:10.1103/PhysRev.85.143.
- Blake, P.; Lee, B.; Summers, M.; Adams, M.; Park, J.-B.; Zhou, Z.; Bax, A. (1992). "Quantitative measurement of small through-hydrogen-bond and 'through-space' 1H–113Cd and 1H–199Hg J couplings in metal-substituted rubredoxin from Pyrococcus furiosus". J. Biomol. NMR. 2 (5): 527–533. doi:10.1007/BF02192814.
- Blake, P. R.; Park, J.-B.; Adams, M. W. W.; Summers, M. F. (1992). "Novel observation of NH–S(Cys) hydrogen-bond-mediated scalar coupling in cadmium-113 substituted rubredoxin from Pyrococcus furiosus". J. Am. Chem. Soc. 114 (12): 4931–4933. doi:10.1021/ja00038a084.
- Dingley, Andrew J.; Cordier, Florence; Grzesiek, Stephan (2001). "An introduction to hydrogen bond scalar couplings". Concepts Magn. Reson. 13 (2): 103–127. doi:10.1002/1099-0534(2001)13:2<103::AID-CMR1001>3.0.CO;2-M.
- Mallory, F. B.; et al. (2000). "Nuclear Spin−Spin Coupling via Nonbonded Interactions. 8. 1 The Distance Dependence of Through-Space Fluorine−Fluorine Coupling". J. Am. Chem. Soc. 122: 4108–4116. doi:10.1021/ja993032z.
- Zong, J.; Mague, J. T.; Kraml, C. M.; Pascal Jr, R. A. (2013). "A Congested in, in-Diphosphine". Org. Lett. 15 (9): 2179–2181. doi:10.1021/ol400728m.
- Zong, J.; Mague, J. T.; Welch, E. C.; Eckert, I. M.; Pascal Jr, R. A. (2013). "Sterically congested macrobicycles with heteroatomic bridgehead functionality". Tetrahedron. 69 (48): 10316–10321. doi:10.1016/j.tet.2013.10.018.