TP53-inducible glycolysis and apoptosis regulator
The TP53-inducible glycolysis and apoptosis regulator (TIGAR) also known as fructose-2,6-bisphosphatase TIGAR is an enzyme that in humans is encoded by the C12orf5 gene.[5][6][7]
TIGAR is a recently discovered enzyme that primarily functions as a regulator of glucose breakdown in human cells. In addition to its role in controlling glucose degradation, TIGAR activity can allow a cell to carry out DNA repair, and the degradation of its own organelles. Finally, TIGAR can protect a cell from death. Since its discovery in 2005 by Kuang-Yu Jen and Vivian G. Cheung, TIGAR has become of particular interest to the scientific community thanks to its active role in many cancers. Normally, TIGAR manufacture by the body is activated by the p53 tumour suppressor protein after a cell has experienced a low level of DNA damage or stress. In some cancers, TIGAR has fallen under the control of other proteins. The hope is that future research into TIGAR will provide insight into new ways to treat cancer.[8][9][10]
This gene is regulated as part of the p53 tumor suppressor pathway and encodes a protein with sequence similarity to the bisphosphate domain of the glycolytic enzyme that degrades fructose-2,6-bisphosphate. The protein functions by blocking glycolysis and directing the pathway into the pentose phosphate shunt. Expression of this protein also protects cells from DNA damaging reactive oxygen species and provides some protection from DNA damage-induced apoptosis. The 12p13.32 region that includes this gene is paralogous to the 11q13.3 region.[7]
Gene
In humans the TIGAR gene, known as C12orf5, is found on chromosome 12p13-3, and consists of 6 exons.[9] The C12orf5 mRNA is 8237 base pairs in length.[11]
Discovery
Jen and Cheung first discovered the c12orf5 gene whilst using computer based searches to find novel p53-regulated genes that were switched on in response to ionizing radiation. They published their research in Cancer Research in 2005.[8]
Later a study focused wholly on the structure and function of the c12orf5 gene was published in Cell by Karim Bensaad et al., in which c12orf5 was given the name TIGAR in honour of its apparent function.[9]
Expression
TIGAR transcription is rapidly activated by the p53 tumour suppressor protein in response to low levels of cellular stress, such as that caused by exposure to low doses of UV.[12] However, under high levels of cellular stress TIGAR expression decreases.[12] P53, a transcription factor, can bind two sites within the human TIGAR gene to activate expression.[9][13] One site is found within the first intron, and binds p53 with high affinity.[9][13] The second is found just prior to the first exon, binds p53 with low affinity,[9][13] and is conserved between mice and humans.[9] TIGAR expression can be regulated by other non-p53 mechanisms in tumour cell lines.[9]
Structure
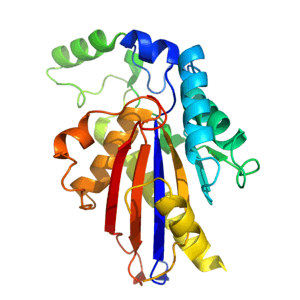
TIGAR is approximately 30kDa [9] and has a tertiary structure that is similar to the histidine phosphatase fold.[14] The core of TIGAR is made up of an α-β-α sandwich, which consists of a six-stranded β sheet surrounded by 4 α helices.[14] Additional α helices and a long loop are built around the core to give the full enzyme.[14] TIGAR has an active site that is structurally similar to that of PhoE (a bacterial phosphatase enzyme) and functionally similar to that of fructose-2,6-bisphosphatase.
The bis-phosphatase-like active site of TIGAR is positively charged, and catalyses the removal of phosphate groups from other molecules.[9][14] In contrast to Fructose-2,6-Bisphosphatase, TIGAR's active site is open and accessible like that of PhoE.[14] The site contains 3 crucial amino acids (2 histidines and 1 glutamic acid [9]) that are involved in the phosphatase reaction. These 3 residues are known collectively as a catalytic triad,[9] and are found in all enzymes belonging to the phosphoglyceromutase branch of the histidine phosphatase superfamily.[9][14] One of the histidine residues is electrostatically bound to a negatively charged phosphate. A second phosphate is bound elsewhere in the active site.[14]
Function
TIGAR activity can have multiple cellular effects. TIGAR acts as a direct regulator of fructose-2,6-bisphosphate levels and hexokinase 2 activity, and this can lead indirectly to many changes within the cell in a chain of biochemical events. TIGAR is a fructose bisphosphatase which activates p53, in results of inhibiting the expression of glucose transporter and also regulating the expression of hexokinase and phosphoglycerate mutase. TIGAR also inhibit the Phosphofructokinase (PFK) by lowering the level of fructose-2,6,bisphosphate, therefore, glycolysis is inhibited and pentose phosphate pathway is promoted.[15]
Fructose-2,6-bisphosphate regulation
TIGAR decreases cellular fructose-2,6-bisphosphate levels.[9][13] It catalyses the removal of a phosphate group from fructose-2,6-bisphosphate (F-2,6-BP):[9][13] Fructose-2,6-Bisphosphate->Fructose-6-phosphate (F-6-P) + phosphate
F-2,6-BP is an allosteric regulator of cellular glucose metabolism pathways. Ordinarily F-2,6-BP binds to and increases the activity of phosphofructokinase 1. Phosphofructokinase-1 catalyses the addition of a phosphate to F-6-P to form Fructose-1,6-bisphosphate (F-1,6-BP). This is an essential step in the glycolysis pathway, which forms the first part of aerobic respiration in mammals.F-2,6-BP also binds to and decreases the activity of fructose-1,6-bisphosphatase.[9] Fructose-1,6-bisphosphatase catalyses the removal of phosphate from F-1,6-BP to form F-6-P. This reaction is part of the gluconeogenesis pathway, which synthesizes glucose, and is the reverse of glycolysis.[16] When TIGAR decreases F-2,6-BP levels, phosphofructokinase becomes less active whilst fructose-1,6-bisphosphatase activity increases.[9][13] Fructose-6-phosphate levels build up,[9][13] which has multiple effects inside the cell:
- The rate of glycolysis decreases[9][13]
- The rate of gluconeogenesis increases[9]
- Excess fructose-6-phosphate is converted to glucose-6-phosphate in an isomerization reaction [9]
- Excess glucose-6-phosphate enters the pentose phosphate pathway. This ultimately leads to the removal of reactive oxygen species (ROS) in the cell[9][13]
- The removal of ROS helps to prevent apoptosis (cell suicide), and may also reduce build-up of DNA damage over time.[9][13]
DNA damage response and cell cycle arrest
TIGAR can act to prevent a cell progressing through the stages of its growth and division cycle by decreasing cellular ATP levels.[12] This is known as cell cycle arrest.[12] This function of TIGAR forms part of the p53 mediated DNA damage response where, under low levels of cellular stress, p53 initiates cell cycle arrest to allow the cell time for repair.[13][17][18] Under high levels of cellular stress, p53 initiates apoptosis instead.[13][17][18]
In non-resting cells, the cell cycle consists of G0 -> G1 -> S -> G2 -> M phases, and is tightly regulated at checkpoints between the phases.[19] If the cell has undergone stress, certain proteins are expressed that will prevent the specific sequence of macromolecular interactions at the checkpoint required for progression to the next phase.[17][18][19]
TIGAR activity can prevent cells progressing into S phase through a checkpoint known in humans as the restriction point. At the very start of G1 phase, a protein called retinoblastoma (Rb) exists in an un-phosphorylated state. In this state, Rb binds to a protein transcription factor E2F and prevents E2F from activating transcription of proteins essential for S-phase. During a normal cell cycle, as G1 progresses, Rb will become phosphorylated in a specific set of sequential steps by proteins called cyclin dependent kinases (cdks) bound to cyclin proteins. The specific complexes that phosphorylate Rb are cyclin D-cdk4 and cyclin E-cdk2.[20]
When Rb has been phosphorylated many times, it dissociates from E2F. E2F is free to activate expression of S-phase genes.[20] TIGAR can indirectly prevent a cell passing through the Restriction Point by keeping Rb unphosphorylated.[12]
When expressed, TIGAR decreases cellular ATP levels through its phosphatase activity.[12] Less ATP is available for Rb phosphorylation, so Rb remains un-phosphorylated and bound to E2F, which cannot activate S phase genes.[12] Expression of cyclin D, ckd4, cyclin E and cdk2 decreases when TIGAR is active, due to a lack of ATP essential for their transcription and translation.[12] This TIGAR activity serves to arrest cells in G1.[12]
Activity of hexokinase 2
Under low oxygen conditions known as hypoxia, a small amount of TIGAR travels to the mitochondria and increases the activity of Hexokinase 2 (HK2) by binding to it[21]
During hypoxia, a protein called Hif1α is activated and causes TIGAR to re-localise from the cytoplasm to the outer mitochondrial membrane.[21] Here, HK2 is bound to an anion channel in the outer mitochondrial membrane called VDAC.[22] TIGAR binds hexokinase 2 and increases its activity by an as yet unknown mechanism.[21]
Hexokinase 2 (HK2) carries out the following reaction:
Glucose + ATP -> Glucose-6-phosphate + ADP [21][22][23]
HK2 is believed to maintain the mitochondrial membrane potential by keeping ADP levels high.[23] It also prevents apoptosis in several ways: it reduces mitochondrial ROS levels,[21][23] and it prevents apoptosis-causing protein Bax from creating a channel with VDAC.[22] This stops cytochrome C protein passing out through VDAC into the cytoplasm where it triggers apoptosis via a caspase protein cascade.[22]
TIGAR does not re-localise to the mitochondria and bind HK2 under normal cellular conditions,[21] or if the cell is starved of glucose.[21] Re-localisation to the mitochondria does not require TIGAR’s phosphatase domain.[21] Instead 4 amino acids at the C-terminal end of TIGAR are essential.[21]
Protection from apoptosis
Increased expression of TIGAR protects cells from oxidative-stress induced apoptosis [24] by decreasing the levels of ROS.[9] TIGAR can indirectly reduce ROS in two distinctive ways. The intracellular environment of the cell will determine which of these two modes of TIGAR action is more prevalent in the cell at any one time.[9][21]
The fructose-2,6-bisphosphatase activity of TIGAR reduces ROS by increasing the activity of the Pentose Phosphate Pathway (PPP).[9] Glucose-6-phosphate builds up due to de-phosphorylation of F-2,6-BP by TIGAR and enters the PPP.[9] This causes the PPP to generate more nicotinamide adenine dinucleotide (NADPH).[9][25] NADPH is a carrier of electrons that is used by the cell as a reducing agent in many anabolic reactions. NADPH produced by the PPP passes electrons to an oxidized glutathione molecule (GSSG) to form reduced glutathione (GSH).[9][25]
GSH becomes the reducing agent, and passes electrons on to the ROS hydrogen peroxide to form harmless water in the reaction:
GSH + H202 -> H20 + GSSG [9][25]
The decrease in H202 as a result of TIGAR activity protects against apoptosis.[9][25]
TIGAR also reduces ROS by increasing the activity of HK2. HK2 reduces ROS levels indirectly by keeping ADP levels at the outer mitochondrial membrane high. If ADP levels fall, the rate of respiration decreases and causes the electron transport chain to become over-reduced with excess electrons. These excess electrons pass to oxygen and form ROS.[21]
The action of the TIGAR/HK2 complex only protects cells from apoptosis under low oxygen conditions. Under normal or glucose starved conditions, TIGAR mediated protection from apoptosis comes from its bis-phosphatase activity alone.[21]
TIGAR cannot prevent apoptosis via death pathways that are independent from ROS and p53.[9] In some cells, TIGAR expression can push cells further towards apoptosis.[9]
Interleukin 3 (IL-3) is a growth factor that can bind to receptors on a cell’s surface and tells the cell to survive and grow.[26] When IL-3 dependent cell lines are deprived of IL-3 they die[26] due to decreased uptake and metabolism of glucose.[26] When TIGAR is overexpressed in IL-3 deprived cells the rate of glycolysis decreases further which enhances the apoptosis rate.[9]
Autophagy
Autophagy is when a cell digests some of its own organelles by lysosomal degradation. Autophagy is employed to remove damaged organelles, or under starvation conditions to provide additional nutrients. Normally, autophagy occurs by the TSC-Mtor pathway, but can be induced by ROS. TIGAR, even at very low levels, inhibits autophagy by decreasing ROS levels. The mechanism by which TIGAR does this is independent from the Mtor pathway, but the exact details are unknown.[27]
Possible roles in cancer
TIGAR can promote development or inhibition of several cancers depending on the cellular context.[13][28][29][30] TIGAR can have some effect on three characteristics of cancer; the ability to evade apoptosis, uncontrolled cell division, and altered metabolism.[13][28][29][30][31] Many cancer cells have altered metabolism where the rate of glycolysis and anaerobic respiration are very high whilst oxidative respiration is low, which is called the Warburg Effect (or aerobic glycolysis).[31] This allows cancer cells to survive under low oxygen conditions, and use molecules from respiratory pathways to synthesise amino acids and nucleic acids to maintain rapid growth.[31]
In Glioma, a type of brain cancer, TIGAR can be over-expressed where it has oncogenic-like effects.[28] In this case, TIGAR acts to maintain energy levels for increased growth by increasing respiration (conferring altered metabolism), and also protects glioma cells against hypoxia-induced apoptosis by decreasing ROS (conferring evasion of apoptosis).[28] TIGAR is also overexpressed in some breast cancers.[30]
In multiple myeloma, TIGAR expression is linked to the activity of MUC-1. MUC-1 is an oncoprotein that is overexpressed in multiple myeloma and protects these cells from ROS-induced apoptosis by maintaining TIGAR activity. When MUC-1 activity is removed, levels of TIGAR decline and cells undergo ROS-induced apoptosis.[29]
In a type of head and neck cancer known as nasopharyngeal cancer, the onco-protein kinase c-Met maintains TIGAR expression. TIGAR increases glycolytic rate and NADPH levels which allows the cancer cells to maintain fast growth rates.[32]
However, TIGAR may also have an inhibitory effect on cancer development by preventing cellular proliferation through its role in p53 -mediated cell cycle arrest.[13]
References
- GRCh38: Ensembl release 89: ENSG00000078237 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000038028 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Jen KY, Cheung VG (Sep 2005). "Identification of novel p53 target genes in ionizing radiation response". Cancer Res. 65 (17): 7666–73. doi:10.1158/0008-5472.CAN-05-1039. PMID 16140933.
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH (Jul 2006). "TIGAR, a p53-inducible regulator of glycolysis and apoptosis". Cell. 126 (1): 107–20. doi:10.1016/j.cell.2006.05.036. PMID 16839880.
- "Entrez Gene: C12orf5 chromosome 12 open reading frame 5".
- Jen KY, Cheung VG (September 2005). "Identification of novel p53 target genes in ionizing radiation response". Cancer Res. 65 (17): 7666–73. doi:10.1158/0008-5472.CAN-05-1039. PMID 16140933.
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH (July 2006). "TIGAR, a p53-inducible regulator of glycolysis and apoptosis". Cell. 126 (1): 107–20. doi:10.1016/j.cell.2006.05.036. PMID 16839880.
- Green DR, Chipuk JE (July 2006). "p53 and metabolism: Inside the TIGAR". Cell. 126 (1): 30–2. doi:10.1016/j.cell.2006.06.032. PMID 16839873.
- "NCBI Summary C12orf5 chromosome 12 open reading frame 5".
- Madan E, Gogna R, Kuppusamy P, Bhatt M, Pati U, Mahdi AA (July 2012). "TIGAR induces p53-mediated cell-cycle arrest by regulation of RB-E2F1 complex". Br. J. Cancer. 107 (3): 516–26. doi:10.1038/bjc.2012.260. PMC 3405207. PMID 22782351.
- Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA (December 2011). "Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor". Oncotarget. 2 (12): 948–57. doi:10.18632/oncotarget.389. PMC 3282098. PMID 22248668.
- Li H, Jogl G (January 2009). "Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator)". J. Biol. Chem. 284 (3): 1748–54. doi:10.1074/jbc.M807821200. PMC 2615519. PMID 19015259.
- Garrett, Reginald (2016). Biochemistry. Stamford, CT: Cengage Learning. p. 631. ISBN 978-1305577206.
- Voet JG, Voet D (2004). Biochemistry (3 ed.). New York: J. Wiley & Sons. ISBN 0-471-19350-X.
- Vousden KH, Lu X (August 2002). "Live or let die: the cell's response to p53". Nat. Rev. Cancer. 2 (8): 594–604. doi:10.1038/nrc864. PMID 12154352.
- Jackson SP, Bartek J (October 2009). "The DNA-damage response in human biology and disease". Nature. 461 (7267): 1071–8. Bibcode:2009Natur.461.1071J. doi:10.1038/nature08467. PMC 2906700. PMID 19847258.
- Elledge SJ (December 1996). "Cell cycle checkpoints: preventing an identity crisis". Science. 274 (5293): 1664–72. Bibcode:1996Sci...274.1664E. doi:10.1126/science.274.5293.1664. PMID 8939848.
- Henley SA, Dick FA (2012). "The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle". Cell Div. 7 (1): 10. doi:10.1186/1747-1028-7-10. PMC 3325851. PMID 22417103.
- Cheung EC, Ludwig RL, Vousden KH (December 2012). "Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death". Proc. Natl. Acad. Sci. U.S.A. 109 (50): 20491–6. Bibcode:2012PNAS..10920491C. doi:10.1073/pnas.1206530109. PMC 3528527. PMID 23185017.
- Pastorino JG, Shulga N, Hoek JB (March 2002). "Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis". J. Biol. Chem. 277 (9): 7610–8. doi:10.1074/jbc.M109950200. PMID 11751859.
- da-Silva WS, Gómez-Puyou A, de Gómez-Puyou MT, Moreno-Sanchez R, De Felice FG, de Meis L, Oliveira MF, Galina A (September 2004). "Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria". J. Biol. Chem. 279 (38): 39846–55. doi:10.1074/jbc.M403835200. PMID 15247300.
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007). "Free radicals and antioxidants in normal physiological functions and human disease". Int. J. Biochem. Cell Biol. 39 (1): 44–84. doi:10.1016/j.biocel.2006.07.001. PMID 16978905.
- Fico A, Paglialunga F, Cigliano L, Abrescia P, Verde P, Martini G, Iaccarino I, Filosa S (August 2004). "Glucose-6-phosphate dehydrogenase plays a crucial role in protection from redox-stress-induced apoptosis". Cell Death Differ. 11 (8): 823–31. doi:10.1038/sj.cdd.4401420. PMID 15044966.
- Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB (September 2001). "Growth factors can influence cell growth and survival through effects on glucose metabolism". Mol. Cell. Biol. 21 (17): 5899–912. doi:10.1128/MCB.21.17.5899-5912.2001. PMC 87309. PMID 11486029.
- Bensaad K, Cheung EC, Vousden KH (October 2009). "Modulation of intracellular ROS levels by TIGAR controls autophagy". EMBO J. 28 (19): 3015–26. doi:10.1038/emboj.2009.242. PMC 2736014. PMID 19713938.
- Wanka C, Steinbach JP, Rieger J (September 2012). "Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis". J. Biol. Chem. 287 (40): 33436–46. doi:10.1074/jbc.M112.384578. PMC 3460445. PMID 22887998.
- Yin L, Kosugi M, Kufe D (January 2012). "Inhibition of the MUC1-C oncoprotein induces multiple myeloma cell death by down-regulating TIGAR expression and depleting NADPH". Blood. 119 (3): 810–6. doi:10.1182/blood-2011-07-369686. PMC 3265204. PMID 22117045.
- Won KY, Lim SJ, Kim GY, Kim YW, Han SA, Song JY, Lee DK (February 2012). "Regulatory role of p53 in cancer metabolism via SCO2 and TIGAR in human breast cancer". Hum. Pathol. 43 (2): 221–8. doi:10.1016/j.humpath.2011.04.021. PMID 21820150.
- Hanahan D, Weinberg RA (March 2011). "Hallmarks of cancer: the next generation". Cell. 144 (5): 646–74. doi:10.1016/j.cell.2011.02.013. PMID 21376230.
- Lui VW, Wong EY, Ho K, Ng PK, Lau CP, Tsui SK, Tsang CM, Tsao SW, Cheng SH, Ng MH, Ng YK, Lam EK, Hong B, Lo KW, Mok TS, Chan AT, Mills GB (March 2011). "Inhibition of c-Met downregulates TIGAR expression and reduces NADPH production leading to cell death". Oncogene. 30 (9): 1127–34. doi:10.1038/onc.2010.490. PMC 3428712. PMID 21057531.
Further reading
- Green DR, Chipuk JE (July 2006). "p53 and metabolism: Inside the TIGAR". Cell. 126 (1): 30–2. doi:10.1016/j.cell.2006.06.032. PMID 16839873.
- White, Kenneth E.; Evans, Wayne E.; O'Riordan, Jeffery L.H.; Speer, Marcy C.; Econs, Michael J.; Lorenz-Depiereux, Bettina; Grabowski, Monika; Meitinger, Thomas; Strom, Tim M. (November 2000). "Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23". Nat. Genet. 26 (3): 345–8. doi:10.1038/81664. PMID 11062477.
- Hartley JL, Temple GF, Brasch MA (November 2000). "DNA cloning using in vitro site-specific recombination". Genome Res. 10 (11): 1788–95. doi:10.1101/gr.143000. PMC 310948. PMID 11076863.
- Katoh Y, Katoh M (August 2005). "Comparative genomics on mammalian Fgf6-Fgf23 locus". Int. J. Mol. Med. 16 (2): 355–8. doi:10.3892/ijmm.16.2.355. PMID 16012775.