Biliary atresia
Biliary atresia, also known as extrahepatic ductopenia and progressive obliterative cholangiopathy, is a childhood disease of the liver in which one or more bile ducts are abnormally narrow, blocked, or absent. It can be congenital or acquired. It has an incidence of one in 10,000–15,000 live births in the United States,[2] and a prevalence of one in 16,700 in the British Isles.[3][4] Biliary atresia is most common in East Asia, with a frequency of one in 5,000.
| Extrahepatic biliary atresia | |
|---|---|
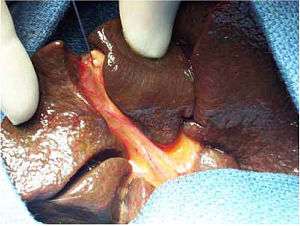 | |
| Intraoperative view of complete extrahepatic biliary atresia[1] | |
| Specialty | Pediatric surgery |
The cause of biliary atresia in Egyptian infants has been proven to be as a result of aflatoxin induced cholangiopathy acquired prenatally in infants who have glutathione S transferase M1 deficiency. The biliary atresia phenotype caused by congenital aflatoxicosis in GST M1 deficient neonates is named Kotb disease.[5] Syndromic biliary atresia (e.g. Biliary Atresia Splenic Malformation (BASM)) has been associated with certain genes (e.g. Polycystic Kidney Disease 1 Like 1 - PKD1L1 [6]) , and some infants with isolated biliary atresia may arise as a result of an autoimmune inflammatory response, possibly due to a viral infection of the liver soon after birth.[7] The only effective treatments[8] are operations such as the Kasai procedure and liver transplantation.[9]
Signs and symptoms
Initially, the symptoms of biliary atresia are indistinguishable from those of neonatal jaundice, a usually harmless condition commonly seen in infants. However, infants with biliary atresia develop progressive conjugated jaundice, pale white stools and dark urine. Some infants fail to thrive as there will be a degree of fat and fat-soluble vitamin malabsorption (e.g. Vitamin K). This may cause a bleeding tendency. Eventually, and usually after 2 months, cirrhosis with portal hypertension will develop. If left untreated, biliary atresia can lead to liver failure. Unlike other forms of jaundice, however, biliary-atresia-related cholestasis mostly does not result in kernicterus, a form of brain damage resulting from liver dysfunction. This is because in biliary atresia, the liver, although diseased, is still able to conjugate bilirubin, and conjugated bilirubin is unable to cross the blood–brain barrier.
Pathophysiology
There are three main types of extra-hepatic biliary atresia:
- Type I: Atresia is restricted to the common bile duct.
- Type II: Atresia of the common hepatic duct.
- Type III: Atresia involves the most proximal part of the bile ducts (>95% of all cases).
In approximately 10% of cases, other anomalies may be associated with biliary atresia. The most common of these syndromic forms is BASM[10] and might include heart lesions, polysplenia, situs inversus, absent venae cavae, and a preduodenal portal vein.[11] Progressive cirrhosis is associated with signs and symptoms of portal hypertension, such as esophagogastric varix bleeding, hypersplenism, hepatorenal syndrome, and hepatopulmonary syndrome.
In an Egyptian study, abnormally high levels of aflatoxin B1 and to a lesser extent aflatoxin B2 was found in liver tissue and blood of all neonates suffering from biliary atresia. Aflatoxins may cause extensive damage to the hepatocytes leading to hepatitis and damage to bile ducts causing inflammation, adhesions and final obstruction of bile ducts.[12] The affected neonates have a genetic detoxification defect that does not allow them to detoxify these aflatoxins timely or effectively. The babies have homozygous deficiency of glutathione S transferase (GST) M1.[13] The aflatoxin damaged liver cells and bile duct cells are removed by neutrophil elastase[14] and by involvement of immune system mediators such as CCL-2 or MCP-1, tumor necrosis factor (TNF), interleukin-6 (IL-6), TGF-beta, endothelin (ET), and nitric oxide (NO). Among these, TGF-beta is the most important pro-fibrogenic cytokine that can be seen in progressive cirrhosis.
The cascade of immune involvement to remove damaged hepatocytes and cholangiocytes ushers regeneration. Yet in infants with biliary atresia regeneration is defective, and results in cirrhosis, as these infants have disrupted p53 and disrupted GSTPi. p53 and GSTPi are responsible for DNA fidelity at regeneration. Hence, these infants get accelerated cirrhosis and march to portal hypertension.[15]
Epidemiology
Biliary atresia seems to affect females slightly more often than males, and Asians and African Americans more often than Caucasians. It is common for only one child in a pair of twins or within the same family to have the condition. There seems to be no link to medications or immunizations given immediately before or during pregnancy. Diabetes during pregnancy particularly during the first trimester seems to predispose to a number of distinct congenital abnormalities in the infant such as sacral agenesis, transposition of the great vessels and the syndromic form of biliary atresia.[16]
Causes
The cause of biliary atresia in most infants is not known and it is likely that a number of factors may play a role. Some may be due to a defect in early bile duct development (particularly those with other abnormalities) and some may arise in the perinatal period due to an external cause such as an hepatotropic virus reovirus 3 infection,[17] congenital cytomegalovirus infection,[18] and autoimmunity.[19] However, experimental evidence is insufficient to confirm any of these theories.[20]
Genetics
An association between biliary atresia and the ADD3 gene was first detected in Chinese populations through a genome-wide association study, and was confirmed in Thai Asians and Caucasians. A possible association with deletion of the gene GPC1, which encodes a glypican 1-a heparan sulfate proteoglycan, has been reported.[21] This gene is located on the long arm of chromosome 2 (2q37) and is involved in the regulation of inflammation and the Hedgehog gene.
Egyptian infants with biliary atresia were found to have null GSTM1 genotype while all their mothers were heterozygous for GSTM1. Thus these infants may be protected in utero by their maternal detoxification system, yet once born they cannot handle the detoxification of an aflatoxin load.
Toxins
Some cases of biliary atresia may result from exposure to aflatoxin B1, and to a lesser extent aflatoxin B2 during late pregnancy. Intact maternal detoxification protects baby during intrauterine life, yet after delivery the baby struggles with the aflatoxin in its blood and liver. Moreover, the baby feeds aflatoxin M1 from its mom, as aflatoxin M1 is the detoxification product of aflatoxin B1. It is a milder toxin that causes cholangitis in the baby.[22]
There are isolated examples of biliary atresia in animals. For instance, lambs born to sheep grazing on land contaminated with a weed (Red Crumbweed) developed biliary atresia at certain times. The plants were later found to contain a toxin, now called biliatresone[23] Studies are ongoing to determine whether there is a link between human cases of biliary atresia and toxins such as biliatresone. There are some indications that a metabolite of certain human gut bacteria may be similar to biliatresone.[24]
Diagnosis
Diagnosis is made by an assessment of history, physical examination in conjunction with blood tests, a liver biopsy, and ultrasound scans imaging and is prompted by prolonged or persistent jaundice, with abnormalities in liver function tests. Ultrasound or other forms of imaging such as radio-isotope liver scans can also be used but final confirmation is usually only reached at the time of exploratory surgery.
Differential diagnoses
The differential diagnoses are extensive and include: Alagille syndrome, alpha-1-antitrypsin deficiency, Byler disease (progressive familial intrahepatic cholestasis), Caroli disease, choledochal cyst, cholestasis, congenital cytomegalovirus disease, congenital herpes simplex virus infection, congenital rubella, congenital syphilis, congenital toxoplasmosis, cystic fibrosis, galactosemia, idiopathic neonatal hepatitis, lipid storage disorders, neonatal hemochromatosis, and total parenteral nutrition-associated cholestasis.[25]
Treatment
Most (>95%) infants with biliary atresia will undergo an operation designed to retain and salvage the native liver, restore bile flow and reduce the level of jaundice. This is known as the Kasai procedure (after Morio Kasai, the Japanese surgeon who first developed the technique) or hepatoportoenterostomy. Although the procedure is not thought of as curative, it may relieve jaundice and stop liver fibrosis, allowing normal growth and development. Published series from Japan, North America and the UK show that bilirubin levels will fall to normal values in about 50-55% of infants, allowing 40-50% to retain their own liver to reach the age of 5 and 10 years (and beyond). Liver transplantation is an option for those children whose liver function and symptoms fail to respond to a Kasai operation.
Recent large-scale studies by Davenport et al. (Annals of Surgery, 2008) show that the age of the patient is not an absolute clinical factor affecting prognosis. The influence of age differs according to the disease etiology—i.e., whether biliary atresia is isolated, cystic (CBA), or accompanied by splenic malformation (BASM).
It is widely accepted that corticosteroid treatment after a Kasai operation, with or without choleretics and antibiotics, has a beneficial effect on postoperative bile flow and can clear jaundice, but the dosing and duration of the ideal steroid protocol are controversial. Furthermore, it has been observed in many retrospective longitudinal studies that corticosteroid treatment does not seem to prolong survival of the native liver or transplant-free survival.
References
- Chardot, Christophe (2006). "Biliary atresia". Orphanet Journal of Rare Diseases. 1: 28. doi:10.1186/1750-1172-1-28. PMC 1560371. PMID 16872500.
- Suchy, Frederick J. (2015). "Anatomy, Histology, Embryology, Developmental Anomalies, and Pediatric Disorders of the Biliary Tract". In Feldman, Mark; Friedman, Lawrence S.; Brandt, Lawrence J. (eds.). Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management (10th ed.). Elsevier Health Sciences. pp. 1055–77. ISBN 978-1-4557-4989-8.
- McKiernan, Patrick J; Baker, Alastair J; Kelly, Deirdre A (2000). "The frequency and outcome of biliary atresia in the UK and Ireland". The Lancet. 355 (9197): 25–9. doi:10.1016/S0140-6736(99)03492-3. PMID 10615887.
- Hartley, Jane L; Davenport, Mark; Kelly, Deirdre A (2009). "Biliary atresia". The Lancet. 374 (9702): 1704–13. doi:10.1016/S0140-6736(09)60946-6. PMID 19914515.
- Kotb MA, Kotb A. Extrahepatic Biliary Atresia is an Aflatoxin Induced Cholangiopathy in Infants with Null GSTM1 Genotype with Disrupted P53 and GSTPi to Mothers Heterozygous for GSTM1 Polymorphism: Damage Control is Mediated through Neutrophil Elastase and CD14+ Activated Monocytes: Kotb Disease http://medicaljournalofcairouniversity.net/home2/images/pdf/2015/march/21.pdf
- Berauer JP, Mezina AI, Okou DT, et al. Identification of Polycystic Kidney Disease 1 Like 1 Gene Variants in Children With Biliary Atresia Splenic Malformation Syndrome. Hepatology. 2019 Sep;70(3):899-910. doi: 10.1002/hep.30515. Epub 2019 Mar 21.
- Mack, Cara (2007). "The Pathogenesis of Biliary Atresia: Evidence for a Virus-Induced Autoimmune Disease". Seminars in Liver Disease. 27 (3): 233–42. doi:10.1055/s-2007-985068. PMC 3796656. PMID 17682970.
- Superina R. Biliary atresia and liver transplantation: results and thoughts for primary liver transplantation in select patients. Pediatr Surg Int. 2017 Dec;33(12):1297-1304. doi: 10.1007/s00383-017-4174-4. Epub 2017 Oct 13. Review. PubMed PMID: 29030698
- Lien, Tien-Hau; Chang, Mei-Hwei; Wu, Jia-Feng; Chen, Huey-Ling; Lee, Hung-Chang; Chen, An-Chyi; Tiao, Mao-Meng; Wu, Tzee-Chung; Yang, Yao-Jong; Lin, Chieh-Chung; Lai, Ming-Wei; Hsu, Hong-Yuan; Ni, Yen-Hsuan (2011). "Effects of the infant stool color card screening program on 5-year outcome of biliary atresia in Taiwan". Hepatology. 53 (1): 202–8. doi:10.1002/hep.24023. PMID 21140377.
- Davenport M, Savage M, Mowat AP, Howard ER. Biliary atresia splenic malformation syndrome: an etiologic and prognostic subgroup. Surgery. 1993 Jun;113(6):662-8.
- Nio, Masaki; Wada, Motoshi; Sasaki, Hideyuki; Tanaka, Hiromu; Watanabe, Tomohiko (2015). "Long-term outcomes of biliary atresia with splenic malformation". Journal of Pediatric Surgery. 50 (12): 2124–7. doi:10.1016/j.jpedsurg.2015.08.040. PMID 26613836.
- Kotb MA. Aflatoxins in Infants with Extrahepatic Biliary Atresia. Med. J. Cairo Univ., Vol. 83, No. 1, March: 207-210, 2015. http://scholar.cu.edu.eg/?q=magdkotb/files/aflatoxins_in_biliary_atresia.pdf
- Kotb MA. Glutathione S Transferase M1 Polymorphism in Extrahepatic Biliary Atresia. Med. J. Cairo Univ., Vol. 83, No. 2, March: 109-112, 2015. http://scholar.cu.edu.eg/?q=magdkotb/files/glutathione_s_transferase_m1_polymorphism_in_extrahepatic_biliary_atresia_.pdf
- Kotb MA. Nuetrophil Elastase Mediated Damage in Infants with Extrahepatic Biliary Atresia: A Prospective Cohort Study.Med. J. Cairo Univ., Vol. 82, No. 2, September: 233-23 7, 2014 http://scholar.cu.edu.eg/?q=magdkotb/files/nuetrophil_elastase_mediated_damage_in_infants_with_extrahepatic_biliary_atresia-_a_prospective_cohort_study_.pdf
- Kotb MA. Evidence of Disruption of p53 and Glutathione S Transferase Pi in Extrahepatic Biliary Atresia in Association with Neutrophil Elastase Mediated Damage. Med. J. Cairo Univ., Vol. 83, No. 1, March: 201-205, 2015. http://scholar.cu.edu.eg/sites/default/files/magdkotb/files/evidence_of_disruption_of_p53_and_glutathione_s_transferase_pi_in_extrahepatic_biliary_atresia_in_association_with_neutrophil_elastase_mediated_damage_.pdf
- Davenport M, Tizzard SA, Underhill J, Mieli-Vergani G, Portmann B, Hadzić N. The biliary atresia splenic malformation syndrome: a 28-year single-center retrospective study. J Pediatr. 2006 Sep;149(3):393-400.
- Mahjoub, Fatemeh; Shahsiah, Reza; Ardalan, Farid; Iravanloo, Guiti; Sani, Mehri; Zarei, Abdolmajid; Monajemzadeh, Maryam; Farahmand, Fatemeh; Mamishi, Setareh (2008). "Detection of Epstein Barr Virus by Chromogenic in Situ Hybridization in cases of extra-hepatic biliary atresia". Diagnostic Pathology. 3: 19. doi:10.1186/1746-1596-3-19. PMC 2424033. PMID 18442403.
- Amer, O. T.; Abd El-Rahma, H. A.; Sherief, L. M.; Hussein, H. F.; Zeid, A. F.; Abd El-Aziz, A. M. (2004). "Role of some viral infections in neonatal cholestasis". The Egyptian Journal of Immunology. 11 (2): 149–55. PMID 16734127.
- Wen, Jie; Xiao, Yongtao; Wang, Jun; Pan, Weihua; Zhou, Ying; Zhang, Xiaoling; Guan, Wenbin; Chen, Yingwei; Zhou, Kejun; Wang, Yang; Shi, Bisheng; Zhou, Xiaohui; Yuan, Zhenghong; Cai, Wei (2014). "Low doses of CMV induce autoimmune-mediated and inflammatory responses in bile duct epithelia of regulatory T cell-depleted neonatal mice". Laboratory Investigation. 95 (2): 180–92. doi:10.1038/labinvest.2014.148. PMID 25531565.
- Saito, Takeshi; Shinozaki, Kuniko; Matsunaga, Tadashi; Ogawa, Tomoko; Etoh, Takao; Muramatsu, Toshinori; Kawamura, Kenji; Yoshida, Hideo; Ohnuma, Naomi; Shirasawa, Hiroshi (2004). "Lack of evidence for reovirus infection in tissues from patients with biliary atresia and congenital dilatation of the bile duct". Journal of Hepatology. 40 (2): 203–11. doi:10.1016/j.jhep.2003.10.025. PMID 14739089.
- Cui, Shuang; Leyva–Vega, Melissa; Tsai, Ellen A.; Eauclaire, Steven F.; Glessner, Joseph T.; Hakonarson, Hakon; Devoto, Marcella; Haber, Barbara A.; Spinner, Nancy B.; Matthews, Randolph P. (2013). "Evidence from Human and Zebrafish That GPC1 is a Biliary Atresia Susceptibility Gene". Gastroenterology. 144 (5): 1107–1115.e3. doi:10.1053/j.gastro.2013.01.022. PMC 3736559. PMID 23336978.
- Kotb MA. Aflatoxins in Infants with Extrahepatic Biliary Atresia. Med. J. Cairo Univ., Vol. 83, No. 1, March: 207-210, 2015. http://scholar.cu.edu.eg/?q=magdkotb/files/aflatoxins_in_biliary_atresia.pdf
- Waisbourd-Zinman, Orith; Koh, Hong; Tsai, Shannon; Lavrut, Pierre-Marie; Dang, Christine; Zhao, Xiao; Pack, Michael; Cave, Jeff; Hawes, Mark; Koo, Kyung A.; Porter, John R.; Wells, Rebecca G. (2016). "The toxin biliatresone causes mouse extrahepatic cholangiocyte damage and fibrosis via decreased glutathione and SOX17". Hepatology. 64 (3): 880–93. doi:10.1002/hep.28599. PMC 4992464. PMID 27081925.
- Patman, Gillian (2015). "Biliary tract: Newly identified biliatresone causes biliary atresia". Nature Reviews Gastroenterology & Hepatology. 12 (7): 369. doi:10.1038/nrgastro.2015.91. PMID 26008130.
- Schwarz SM (25 September 2017). "Pediatric Biliary Atresia Differential Diagnoses". Medscape Pediatrics.
Further reading
- Bhatnagar, V; Kumar, Arun; Gupta, AK (2005). "Choledochal cyst associated with extrahepatic bile duct atresia". Journal of Indian Association of Pediatric Surgeons. 10 (1): 48–9. doi:10.4103/0971-9261.16077.
External links
| Classification | |
|---|---|
| External resources |