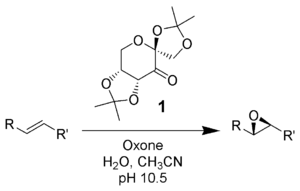
Shi epoxidation
The Shi epoxidation is a chemical reaction described as the asymmetric epoxidation of alkenes with oxone (potassium peroxymonosulfate) and a fructose-derived catalyst (1). This reaction is thought to proceed via a dioxirane intermediate, generated from the catalyst ketone by oxone (potassium peroxymonosulfate). The addition of the sulfate group by the oxone facilitates the formation of the dioxirane by acting as a good leaving group during ring closure. It is notable for its use of a non-metal catalyst and represents an early example of organocatalysis. The reaction was first discovered by Yian Shi (史一安, pinyin: Shǐ Yī-ān) of Colorado State University in 1996.

Brief Historical Background
Many attempts at the synthesis of an efficient non-metal catalyst were made before one was discovered. The problem with previous catalysts was the rapid decomposition/oxidation of the dioxirane intermediate and lack of electrophilicity of the reactive ketone. Aromatic ketones were proposed, and many subsequent variations of oxoammonium salts were used, but were ineffective in promoting epoxidation because of the oxidative instability of the amide groups and high flexibility of the seven-membered rings. Enantioselectivity of these early catalysts were also lowered because of large distances between the asymmetric subunits and reaction centers, yielding less than 10 percent in enantiomeric excess.[1]
The catalyst discovered by Yian Shi's group in 1996 was derived from D-fructose, and has a stereogenic center close to the reacting center (ketone)- the rigid six-membered ring structure of the catalyst and adjacent quaternary ring group minimizes epimerization of this stereocenter. Oxidation by the active dioxirane catalyst takes place from the si-face, due to steric hindrance of the opposing re-face. This catalyst functions efficiently as an asymmetric catalyst for unfunctionalized trans-olefins.[2]
Formation of Dioxirane Catalyst
Under normal pH conditions, an excess of 3 stoichiometric amounts of ketone catalyst are needed due to a high rate of decomposition. At basic pH conditions greater than 10 (pH 10.5) substoichiometric amounts (0.2-0.3) are needed for epoxidations, lowering the decomposition of reagents by disfavoring the Baeyer-Villiger side reaction. Higher temperatures result in further decomposition; thus a low temperature of zero degrees Celsius is used.

Decomposition of reagents is bimolecular (second-order reaction rate), so low amounts of oxone and catalyst are used.
The reaction is mediated by a D-fructose derived catalyst, which produces the (R,R) enantiomer of the resulting epoxide. Solubilities of olefin organic substrate and oxidant (oxone) differ, and thus a biphasic medium is needed. The generation of the active catalyst species takes place in the aqueous layer, and is shuttled to the organic layer with the reactants by tetrabutylammonium sulfate.The ketone catalyst is continuously regenerated in a catalytic cycle, and thus can catalyze the epoxidation in small amounts.
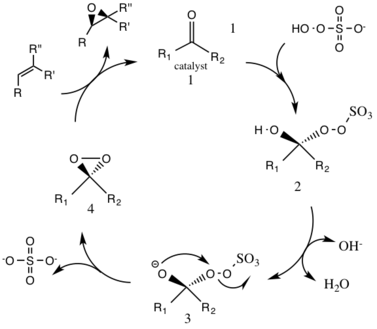
The first step in the catalytic cycle reaction is the nucleophilic addition reaction of the oxone with the ketone group on the catalyst (intermediate 1). This forms the reactive intermediate number 2 species, the Criegee intermediate that can potentially lead to unwanted side reactions, such as the Baeyer-Villiger reaction (see below). The generation of intermediate species number 3 occurs under basic conditions, with a removal of the hydrogen from the hydroxy group to form a nucleophilic oxygen anion. The sulfate group facilitates the subsequent formation of the dioxirane, intermediate species number 4, by acting as a good leaving group during the 3-exo-trig cyclization. The activated dioxirane catalytic species then transfers an oxygen atom to the alkene, leading to a regeneration of the original catalyst.[3]
Side Reactions
A potential side reaction that may occur is the Baeyer-Villiger reaction of intermediate 2, where there is a rearrangement of the peroxy group that results in the formation of the relative ester. The extent of this side reaction declines with the rise of pH, and increases the nucleophilicity of the oxone, making basic conditions favorable for the overall epoxidation and reactivity of the catalytic species.

Mechanism of Epoxidation
The oxygen from the dioxirane group generated on the organic catalyst is transferred to the alkene, in what is thought to be a concerted mechanism, although the presence of an oxygen anion intermediate through an Sn2 mechanism may transpire.
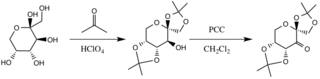
Preparation of D-Fructose Derivative
The catalyst is formed by reaction with acetone under basic conditions, with the hydroxyl groups of the fructose ring acting as nucleophiles, their nucleophilicity increased by the basic conditions created by potassium carbonate. The electron withdrawing substituents (alpha-ether groups) encourage the formation of the ketone from the oxidizing agent pyridinium chlorochromate by increasing the electrophilicity of the carbonyl carbon, via a stabilizing delocalization of the forming π C-C bonds into the σ* C-O bonds of the adjacent ethers.[3]
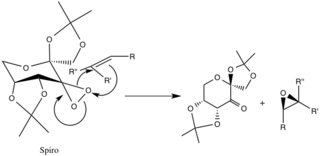
Transition States and Enantiomeric Selectivity
There are two proposed transition states, whose geometries are speculated and not corroborated by experimental evidence, but are attributed to stereoelectronic effects. The spiro transition state is favored over the planar due to the non-bonding orbitals of the superior oxygen donating into the π* anti-bonding C-C orbitals of the reacting alkene, providing a stabilizing delocalization of electrons.
Donation of these electrons into the forming C-O σ bonds of the epoxide bonds also encourages the formation of the spiro-product (the geometry of the product is aligned as well). The planar configuration is disfavored due to lack of pi-backbonding and steric hindrance of the alkyl groups with large alkyl functional groups of the catalytic ring.[4]
The previously mentioned configurations are favored over the transition states of the opposing enantiomers because of unfavorable steric interactions between the R-alkyl groups (see below) and the ether-alkyl functional groups of the catalyst ring.
The enantiomeric success of this epoxidation is relatively high compared to metal catalysts, and generally results in a high enantiomeric excess exceeding 80 percent.[2]
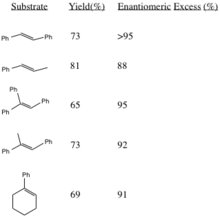
Reaction Yield and Stereoselectivity
This procedure generates epoxides with high enantiomeric excesses from trans-disubstituted alkenes and trisubstituted alkenes. Cis-disubstituted alkenes and styrenes are asymmetrically epoxidized using a similar catalyst. Generation of (R,R) epoxides from corresponding alkenes increases in stereoselectivity with increased steric bulk of substituent R groups (especially in trans-olefins).
References
- Denmark, Scott E. (13 April 1999). "The Development of Chiral, Non-Racemic Dioxiranes for the Catalytic, Enantioselective Epoxidation of Alkenes" (PDF). Synlett. 1999: 847–859. doi:10.1055/s-1999-3123.
- Shi, Yian (1996). "An Efficient Asymmetric Epoxidation Method for trans-olefins mediated by a Fructose-Derived Ketone". Journal of the American Chemical Society. 118 (40): 9806–9807. doi:10.1021/ja962345g.
- "Organic Chemistry Portal".
- Shi, Yian (July 8, 1997). "An Efficient Asymmetric Epoxidation Method". Journal of the American Chemical Society. 119 (46): 11224–11235. doi:10.1021/ja972272g.
- ^ An Efficient Catalytic Asymmetric Epoxidation Method Zhi-Xian Wang, Yong Tu, Michael Frohn, Jian-Rong Zhang, and Yian Shi J. Am. Chem. Soc. 1997, 119(46), 11224-11235. (doi:10.1021/ja972272g)
- ^ Frohn, M.; Shi, Y. Synthesis 2000, 14, 1979-2000 doi:10.1055/s-2000-8715. (Review)
- ^ Tian, H.; She, X.; Shu, L.; Yu, H.; Shi, Y. J. Am. Chem. Soc. 2000, 122, 11551-11552. (doi:10.1021/ja003049d)
- ^ Tian, H.; She, X.; Xu, J.; Shi, Y. Org. Lett. 2001, 3, 1929-1931. (doi:10.1021/ol010066e)
- Shi Epoxidation <https://www.organic-chemistry.org/namedreactions/shi-epoxidation.shtm>
- Denmark, Wu, et al. "The Development of Chiral, Non-Racemic Dioxiranes for the Catalytic, Enantioselective Epoxidation of Alkenes". (13, April 1999) <https://www.thieme-connect.de/products/ejournals/pdf/10.1055/s-1999-3123.pdf>
- Frohn, Shi, Tu, Wang, Zhang, et al. "An Efficient Asymmetric Epoxidation Method". (8 July 1997) <http://pubs.acs.org/doi/pdf/10.1021/ja972272g>
- Shi, Wang, et al. "A New Type of Ketone Catalyst for Asymmetric Epoxidation". (12 September 1997). <http://pubs.acs.org/doi/pdf/10.1021/jo971701q>