Oxidation with dioxiranes
Oxidation with dioxiranes refers to the introduction of oxygen into organic molecules through the action of a dioxirane. Dioxiranes are well known for their oxidation of alkenes to epoxides; however, they are also able to oxidize other unsaturated functionality, heteroatoms, and alkane C-H bonds.[1]
Introduction
Dioxiranes may be produced through the action of KHSO5 on carbonyl compounds. Because of their low-lying σ*O-O orbital, they are highly electrophilic oxidants and react with unsaturated functional groups, Y-H bonds (yielding oxygen insertion products), and heteroatoms.[2] The most common dioxiranes employed for organic synthesis are dimethyldioxirane (DMD) and trifluoromethyl-methyldioxirane (TFD). The latter is effective for chemoselective oxidations of C-H and Si-H bonds.[3] Although this class of reagents is most famous for the epoxidation of alkenes, dioxiranes have been used extensively for other kinds of oxidations as well.
(1)
Mechanism and Stereochemistry
Prevailing Mechanisms
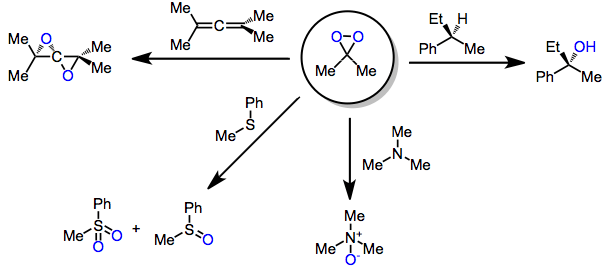
Epoxidations of alkynes and allenes proceed by concerted mechanisms analogous to epoxidations of simple alkenes.[4] Often, these epoxidized products are unstable and undergo further oxidation reactions via different mechanisms, such as Y-H insertion.
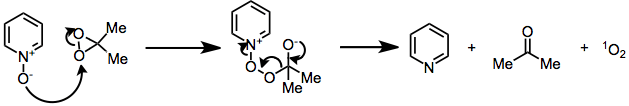
Kinetic studies of heteroatom oxidations have demonstrated that their mechanisms likely proceed by an SN2 process, rather than a single-electron-transfer pathway. An example of heteroatom oxidation is the nucleophilic decomposition of DMD by N-oxides, a side reaction that regenerates the reduced starting material and converts the oxidizing agent to dioxygen and acetone.[5]
(2)
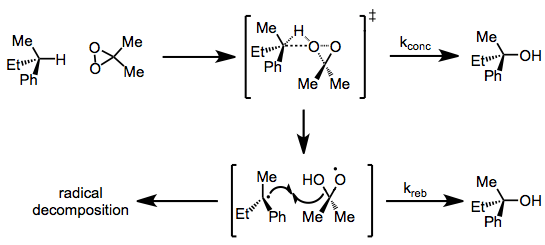
Concerning the mechanism of C-H and Si-H oxidations, two mechanisms have been proposed. The debate centers on whether the oxidation takes place via concerted oxenoid-type insertion or via radical intermediates. A large body of evidence (including analogous oxidations of alkenes and peracid epoxidation) supports the concerted mechanism;[6] however, recent observations of radical reactivity have been made. Complete retention of configuration in oxidations of chiral alkanes rules out the involvement of free, uncaged radicals. However, products of radical decomposition pathways have been observed in some DMD oxidations, suggesting radical intermediates.[7]
(3)
Stereoselective Variants
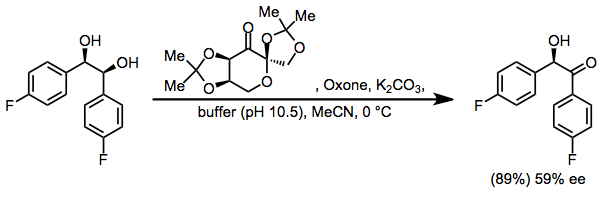
Enantioselective dioxirane oxidations may rely on chiral, non-racemic dioxiranes, such as Shi's fructose-based dioxirane. Enantioselective oxidation of meso-diols with Shi's catalyst, for instance, produces chiral α-hydroxy ketones with moderate enantioselectivity.[8]
(4)
Alternatively, an asymmetric environment may be established through the use of chiral metal complexes. For instance, coordination of sulfides to chiral, non-racemic transition-metal complexes facilitates enantioselective sulfoxidation with dioxiranes. Bovine serum albumin may also be used to establish a chiral environment for sulfoxidation.[9]
(5)
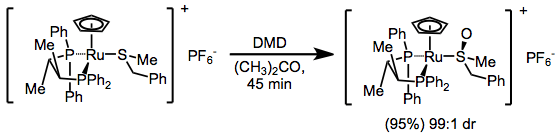
Scope and Limitations
Dioxiranes oxidize a wide variety of functional groups. This section describes the substrate scope of dioxirane epoxidation and the products that most commonly result.
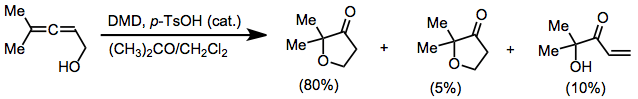
Oxidations of alkynes, allenes, arenes, and other unique unsaturated functionality may yield epoxides or other oxidized products. Oxidation of allenes affords allene dioxides or products of intramolecular participation.[10] Minor amounts of side products derived from additional oxidation or rearrangement were also observed.
(6)
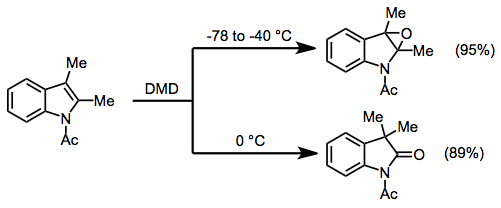
In oxidations of heteroaromatic compounds, the products obtained depend on reaction conditions. Thus, at low temperatures, acetylated indoles are simply epoxidized in high yield (unprotected indoles undergo N-oxidation). However, when the temperature is raised to 0 °C, rearranged products are obtained.[11]
(7)
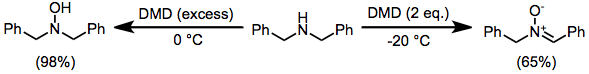
DMD may oxidize heteroatoms to the corresponding oxides (or products of oxide decomposition). Often, the results of these oxidations depend on reaction conditions. Tertiary amines cleanly give the corresponding N-oxides.[12] Primary amines give nitroalkanes upon treatment with 4 equivalents of DMD, but azoxy compounds upon treatment with only 2 equivalents.[13] Secondary amines afford either hydroxylamines or nitrones.[14]
(8)
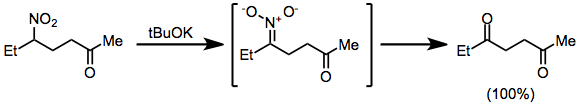
Oxidation of nitronate anions, generated in situ from nitroalkanes, leads to carbonyl compounds in an example of an oxidative Nef reaction.[15]
(9)
Sulfide oxidation in the presence of a single equivalent of DMD leads to sulfoxides.[16] Increasing the amount of DMD used (2 or more equivalents) leads to sulfones. Both nitrogen and sulfur are more susceptible to oxidation than carbon-carbon multiple bonds.
(10)
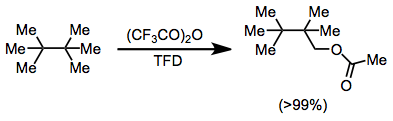
Although alkanes are typically difficult to functionalize directly, C-H insertion with TFD is an efficient process in many cases. The order of reactivity of C-H bonds is: allylic > benzylic > tertiary > secondary > primary. Often, the intermediate alcohols produced are oxidized further to carbonyl compounds, although this can be prevented by trapping in situ with an anhydride. Chiral alkanes are functionalized with retention of configuration.[17]
(11)
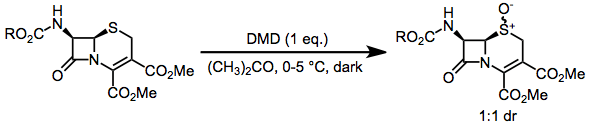
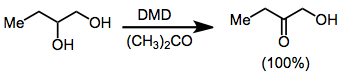
Dioxiranes oxidize primary alcohols to either the aldehyde or carboxylic acid;[18] however, DMD selectively oxidizes secondary over primary alcohols. Thus, vicinal diols may be transformed into α-hydroxy ketones with dioxirane oxidation.[19]
(12)
Epoxidation is usually more facile than C-H oxidation, although sterically hindered allyl groups may undergo selective C-H oxidation instead of epoxidation of the allylic double bond.[20]
Comparison with Other Methods
A variety of alternative heteroatom oxidation reagents are known, including peroxides (often employed with a transition metal catalyst) and oxaziridines. These reagents do not suffer from the over-oxidation problems and decomposition issues associated with dioxiranes; however, their substrate scope tends to be more limited. Nucleophilic decomposition of dioxiranes to singlet oxygen is a unique problem associated with dioxirane heteroatom oxidations. Although chiral dioxiranes do not provide the same levels of enantioselectivity as other protocols, such as Kagan's sulfoxidation system,[21] complexation to a chiral transition metal complex followed by oxidation affords optically active sulfoxides with good enantioselectivity.
Oxidation of arenes and cumulenes leads initially to epoxides. These substrates are resistant to many epoxidation reagents, including oxaziridines, hydrogen peroxide, and manganese oxo compounds. Organometallic oxidants also react sluggishly with these compounds, with the exception of methyltrioxorhenium.[22] Peracids also react with arenes and cumulenes, but cannot be employed with substrates containing acid-sensitive functionality.
The direct oxidative functionalization of C-H bonds is an ongoing problem in oxidation chemistry. Among metal-free systems, dioxiranes are the best oxidants for the conversion of C-H bonds to alcohols or carbonyls. However, some catalytic transition-metal systems, such as White's palladium-sulfoxide system, are able to oxidize C-H bonds selectively.[23]
Experimental Conditions and Procedure
Typical Conditions
Dioxiranes are volatile peroxides and should be handled with care in a well-ventilated fume hood. Inhalation and skin exposure should be strictly avoided. As many dioxirane oxidations depend subtly on reaction conditions, care should also be taken to optimize reaction parameters such as equivalents of oxidant and temperature. Hydroxylic and ethereal solvents should never be used in reactions of dioxiranes, as competitive oxidation of the solvent will take place.
Preparation of TFD[24]
A 150 mL, five-necked round-bottomed vessel is equipped with a mechanical stirrer, an air-cooled straight condenser loosely packed with silanized glass wool, a solids addition funnel, a gas inlet tube extending into the reaction mixture, and a thermometer. The air condenser is connected laterally to the top entry of an efficient-jacketed spiral condenser cooled at −75 °C (dry ice/acetone); the bottom exit of this condenser is fitted to a two-ways fraction-collector adapter carrying two 50 mL receiving flasks, also cooled at −75 °C. In succession to the adapter, two cold traps are placed, the first cooled at −75 °C, and the second at liquid nitrogen temperature. The main vessel is charged with NaHCO3 (1.5 g) and Na2H2EDTA (0.5 g) dissolved in 15 mL of bidistilled water, and cooled at 2–5 °C. To this 20 g (0.179 mol) of 1,1,1-trifluoro-2-propanone is added, and mechanical stirring is initiated. Solid potassium peroxomonosulfate (0.081 mol, 25 g of Curox triple salt) is quickly added (1–2 min) to the reaction vessel, while passing a gentle stream of He (or Ar) gas through the mixture. Just before starting the addition of the solid inorganic peroxide, a slight vacuum (650–700 mmHg) is applied to the last trap. Shortly after the addition is initiated, the end two-ways adaptor is switched to insert the main collection flask, and during 6–8 min approximately 12 mL of yellow solution of TFD in TFP is collected, having dioxirane concentrations ranging from 0.5 to 0.8 M; 1H NMR (CF3COCH3, Me4Si, −20 °C) 6 1.97 (s).
Cold solutions of TFP can be dried briefly over MgSO4 (Analar grade), quickly filtered, and stored over 5Å molecular sieves at −20 °C (keep well protected from light). Provided that care is exercised in using extra clean glassware (before drying, for final wash we use 0.01% Na2H2EDTA in bidistilled water) and that trace metals and other impurities are carefully excluded, only minor loss of dioxirane content of solutions is observed (e.g., 56% during 48 h, at −20 °C). The expensive TFP solvent can be recovered from decomposed dioxirane solutions by low-temperature fractional distillation (bp 22 °C) through an efficient column.
Example Oxidation Procedure[24]
(13)
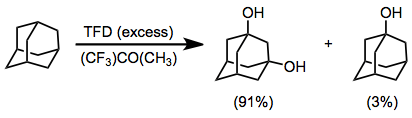
A solution of TFD (4.60 mL, 0.50 M, 2.30 mmol) in trifluoroacetone/CH2Cl2 (2:1 v/v) at –20 ° was added to a solution of adamantane (136 mg, 0.100 mmol) in CH2Cl2 (5 mL) also at –20 °, while stirring vigorously magnetically. The progress of the reaction was followed by GLC analysis, which indicated that 97% of the adamantane was converted to its hydroxylated products in 40 minutes. Removal of the solvent on a rotary evaporator (–20 °, 15 mmHg) afforded a mixture of the 1,3-dihydroxyadamantane (156 mg, 91%) and the monohydroxy adamantane (4.6 mg, 3%). Spectral data for 1,3-dihydroxyadamantane is as follows. IR (KBr): 3275 (O-H), 2930 and 2851 (C-H), 1453, 1135, 1027, 968, 911, 801 cm−1. 1H NMR (CDCl3): 1.50 (m, 2 H), 1.62 (apparent d, 8 H), 1.65 (s, 2 H), 2.23 (m, 2 H), 4.88 (OH).
References
- Adam, W.; Zhao, C.-G.; Jakka, K. Org. React. 2007, 69, 1. doi:10.1002/0471264180.or069.01
- Adam, W.; Curci, R.; Edwards, J. O. Acc. Chem. Res. 1989, 22, 205.
- Asensio, G.; González-Núñez, M. E.; Biox Bernardini, C.; Mello, R.; Adam, W. J. Am. Chem. Soc. 1993, 115, 7250.
- Adam, W.; Saha-Moller, C.; Jakka, K. Org. React. 2002, 61, 219.
- Adam, W.; Briviba, K.; Duschek, F.; Golsch, D.; Kiefer, W.; Sies, H. J. Chem. Soc., Chem. Commun. 1995, 1831.
- Mello, R.; Fiorentino, M.; Fusco, C.; Curci, R. J. Am. Chem. Soc. 1989, 111, 6749.
- Bravo, A.; Fontana, F.; Fronza, G.; Minisci, F.; Zhao, L. J. Org. Chem. 1998, 63, 254.
- Adam, W.; Saha-Möller, C. R.; Zhao, C.-G. J. Org. Chem. 1999, 64, 7492.
- Schenk, W. A.; Steinmetz, B.; Hagel, M.; Adam, W.; Saha-Möller, C. R. Z. Naturforsch. 1997, 1359.
- Crandall, J. K.; Batal, D. J.; Lin, F.; Reix, T.; Nadol, G. S.; Ng, R. A. Tetrahedron 1992, 48, 1427.
- Adam, W.; Ahrweiler, M.; Peters, K.; Schmiedeskamp, B. J. Org. Chem. 1994, 59, 2733.
- Ferrer, M.; Sánchez-Baeza, F.; Messeguer, A. Tetrahedron 1997, 53, 15877.
- Murray, R. W.; Singh, M.; Rath, N. Tetrahedron: Asymmetry 1996, 7, 1611.
- Murray, R. W.; Singh, M.; Jeyaraman, R. J. Am. Chem. Soc. 1992, 114, 1346.
- Pinnick, H. W. Org. React. 1990, 38, 655.
- Murray, R. W.; Jeyaraman, R. J. Org. Chem. 1985, 50, 2847.
- Asensio, G.; Mello, R.; González-Núñez, M. E.; Castellano, G.; Corral, J. Angew. Chem. Int. Ed. Engl. 1996, 35, 217.
- Curci, R. J. Am. Chem. Soc. 1991, 113, 2205.
- Bovicelli, P.; Lupattelli, P.; Sanetti, A.; Mincione, E. Tetrahedron Lett. 1994, 35, 8477.
- Adam, W.; Prechtl, F.; Richter, M. J.; Smerz, A. K. Tetrahedron Lett. 1995, 36, 4991.
- Pitchen, P.; Dunach, E.; Deshmukh, M. N.; Kagan, H. B. J. Am. Chem. Soc. 1984, 106, 8188.
- Adam, W.; Mitchell, C. M.; Saha-Möller, C. R.; Weichold, O. In Structure and Bonding, Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations; Meunier, B., Ed.; Springer Verlag: Berlin Heidelberg, 2000; Vol. 97, pp 237–285.
- N.A. Vermeulen; M.S. Chen; and M.C. White. Tetrahedron 2009, 65, 3078.
- Mello, R.; Cassidei, L.; Fiorentino, M.; Fusco, C.; Curci, R. Tetrahedron Lett. 1990, 31, 3067.