Sequencing
In genetics and biochemistry, sequencing means to determine the primary structure (sometimes incorrectly called the primary sequence) of an unbranched biopolymer. Sequencing results in a symbolic linear depiction known as a sequence which succinctly summarizes much of the atomic-level structure of the sequenced molecule.
DNA sequencing
DNA sequencing is the process of determining the nucleotide order of a given DNA fragment. So far, most DNA sequencing has been performed using the chain termination method developed by Frederick Sanger. This technique uses sequence-specific termination of a DNA synthesis reaction using modified nucleotide substrates. However, new sequencing technologies such as pyrosequencing are gaining an increasing share of the sequencing market. More genome data are now being produced by pyrosequencing than Sanger DNA sequencing. Pyrosequencing has enabled rapid genome sequencing. Bacterial genomes can be sequenced in a single run with several times coverage with this technique. This technique was also used to sequence the genome of James Watson recently.[1]
The sequence of DNA encodes the necessary information for living things to survive and reproduce. Determining the sequence is therefore useful in fundamental research into why and how organisms live, as well as in applied subjects. Because of the key importance DNA has to living things, knowledge of DNA sequences is useful in practically any area of biological research. For example, in medicine it can be used to identify, diagnose, and potentially develop treatments for genetic diseases. Similarly, research into pathogens may lead to treatments for contagious diseases. Biotechnology is a burgeoning discipline, with the potential for many useful products and services.
The Carlson curve is a term coined by The Economist [2] to describe the biotechnological equivalent of Moore's law, and is named after author Rob Carlson.[3] Carlson accurately predicted the doubling time of DNA sequencing technologies (measured by cost and performance) would be at least as fast as Moore's law.[4] Carlson curves illustrate the rapid (in some cases hyperexponential) decreases in cost, and increases in performance, of a variety of technologies, including DNA sequencing, DNA synthesis, and a range of physical and computational tools used in protein expression and in determining protein structures.
Sanger sequencing
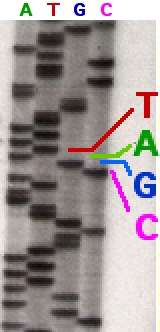
In chain terminator sequencing (Sanger sequencing), extension is initiated at a specific site on the template DNA by using a short oligonucleotide 'primer' complementary to the template at that region. The oligonucleotide primer is extended using a DNA polymerase, an enzyme that replicates DNA. Included with the primer and DNA polymerase are the four deoxynucleotide bases (DNA building blocks), along with a low concentration of a chain terminating nucleotide (most commonly a di-deoxynucleotide). The deoxynucleotides lack in the OH group both at the 2' and at the 3' position of the ribose molecule, therefore once they are inserted within a DNA molecule they prevent it from being further elongated. In this sequencer four different vessels are employed, each containing only of the four dideoxyribonucleotides; the incorporation of the chain terminating nucleotides by the DNA polymerase in a random position results in a series of related DNA fragments, of different sizes, that terminate with a given dideoxiribonucleotide. The fragments are then size-separated by electrophoresis in a slab polyacrylamide gel, or more commonly now, in a narrow glass tube (capillary) filled with a viscous polymer.
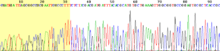
An alternative to the labelling of the primer is to label the terminators instead, commonly called 'dye terminator sequencing'. The major advantage of this approach is the complete sequencing set can be performed in a single reaction, rather than the four needed with the labeled-primer approach. This is accomplished by labelling each of the dideoxynucleotide chain-terminators with a separate fluorescent dye, which fluoresces at a different wavelength. This method is easier and quicker than the dye primer approach, but may produce more uneven data peaks (different heights), due to a template dependent difference in the incorporation of the large dye chain-terminators. This problem has been significantly reduced with the introduction of new enzymes and dyes that minimize incorporation variability. This method is now used for the vast majority of sequencing reactions as it is both simpler and cheaper. The major reason for this is that the primers do not have to be separately labelled (which can be a significant expense for a single-use custom primer), although this is less of a concern with frequently used 'universal' primers. This is changing rapidly due to the increasing cost-effectiveness of second- and third-generation systems from Illumina, 454, ABI, Helicos, and Dover.
Pyrosequencing
The pyrosequencing method is based on the detection of the pyrophosphate release on nucleotide incorporation. Before performing pyrosequencing, the DNA strand to sequence has to be amplified by PCR. Then the order in which the nucleotides have to be added in the sequencer is chosen (i.e. G-A-T-C). When a specific nucleotide is added, if the DNA polymerase incorporates it in the growing chain, the pyrophosphate is released and converted into ATP by ATP sulfurylase. ATP powers the oxidation of luciferase through the luciferase; this reaction generates a light signal recorded as a pyrogram peak. In this way, the nucleotide incorporation is correlated to a signal. The light signal is proportional to the amount of nucleotides incorporated during the synthesis of the DNA strand (i.e. two nucleotides incorporated correspond to two pyrogram peaks). When the added nucleotides aren't incorporated in the DNA molecule, no signal is recorded; the enzyme apyrase removes any unincorporated nucleotide remaining in the reaction. This method requires neither fluorescently-labelled nucleotides nor gel electrophoresis. Pyrosequencing, which was developed by Pål Nyrén and Mostafa Ronaghi DNA, has been commercialized by Biotage (for low-throughput sequencing) and 454 Life Sciences (for high-throughput sequencing). The latter platform sequences roughly 100 megabases [now up to 400 megabases] in a seven-hour run with a single machine. In the array-based method (commercialized by 454 Life Sciences), single-stranded DNA is annealed to beads and amplified via EmPCR. These DNA-bound beads are then placed into lls on a fiber-optic chip along with enzymes which produce light in the presence of ATP. When free nucleotides are washed over this chip, light is produced as ATP is generated when nucleotides join with their complementary base pairs. Addition of one (or more) nucleotide(s) results in a reaction that generates a light signal that is recorded by the CCD camera in the instrument. The signal strength is proportional to the number of nucleotides, for example, homopolymer stretches, incorporated in a single nucleotide flow.
True single molecule sequencing
Large-scale sequencing
Whereas the methods above describe various sequencing methods, separate related terms are used when a large portion of a genome is sequenced. Several platforms were developed to perform exome sequencing (a subset of all DNA across all chromosomes that encode genes) or whole genome sequencing (sequencing of the all nuclear DNA of a human).
RNA sequencing
RNA is less stable in the cell, and also more prone to nuclease attack experimentally. As RNA is generated by transcription from DNA, the information is already present in the cell's DNA. However, it is sometimes desirable to sequence RNA molecules. While sequencing DNA gives a genetic profile of an organism, sequencing RNA reflects only the sequences that are actively expressed in the cells. To sequence RNA, the usual method is first to reverse transcribe the RNA extracted from the sample to generate cDNA fragments. This can then be sequenced as described above. The bulk of RNA expressed in cells are ribosomal RNAs or small RNAs, detrimental for cellular translation, but often not the focus of a study. This fraction can be removed in vitro, however, to enrich for the messenger RNA, also included, that usually is of interest. Derived from the exons these mRNAs are to be later translated to proteins that support particular cellular functions. The expression profile therefore indicates cellular activity, particularly desired in the studies of diseases, cellular behaviour, responses to reagents or stimuli. Eukaryotic RNA molecules are not necessarily co-linear with their DNA template, as introns are excised. This gives a certain complexity to map the read sequences back to the genome and thereby identify their origin. For more information on the capabilities of next-generation sequencing applied to whole transcriptomes see: RNA-Seq and MicroRNA Sequencing.
Protein sequencing
Methods for performing protein sequencing include:
- Edman degradation
- Peptide mass fingerprinting
- Mass spectrometry
- Protease digests
If the gene encoding the protein is known, it is currently much easier to sequence the DNA and infer the protein sequence. Determining part of a protein's amino-acid sequence (often one end) by one of the above methods may be sufficient to identify a clone carrying this gene.
Polysaccharide sequencing
Though polysaccharides are also biopolymers, it is not so common to talk of 'sequencing' a polysaccharide, for several reasons. Although many polysaccharides are linear, many have branches. Many different units (individual monosaccharides) can be used, and bonded in different ways. However, the main theoretical reason is that whereas the other polymers listed here are primarily generated in a 'template-dependent' manner by one processive enzyme, each individual join in a polysaccharide may be formed by a different enzyme. In many cases the assembly is not uniquely specified; depending on which enzyme acts, one of several different units may be incorporated. This can lead to a family of similar molecules being formed. This is particularly true for plant polysaccharides. Methods for the structure determination of oligosaccharides and polysaccharides include NMR spectroscopy and methylation analysis.[5]
See also
- Exome sequencing
- Full genome sequencing
- Genetic code
- Pathogenomics
- RNA-Seq
- MicroRNA sequencing
- Sequence motif
References
- Wheeler, David A.; Srinivasan, Maithreyan; Egholm, Michael; Shen, Yufeng; Chen, Lei; McGuire, Amy; He, Wen; Chen, Yi-Ju; Makhijani, Vinod (2008-04-17). "The complete genome of an individual by massively parallel DNA sequencing". Nature. 452 (7189): 872–876. Bibcode:2008Natur.452..872W. doi:10.1038/nature06884. ISSN 0028-0836. PMID 18421352.
- Life 2.0. (2006, August 31). The Economist
- Carlson, Robert H. Biology Is Technology: The Promise, Peril, and New Business of Engineering Life. Cambridge, MA: Harvard UP, 2010. Print
- Carlson, Robert (2003). "The Pace and Proliferation of Biological Technologies". Biosecurity and Bioterrorism: Biodefense Strategy, Practice, and Science. 1 (3): 203–214. doi:10.1089/153871303769201851. PMID 15040198.
- A practical guide to structural analysis of carbohydrates