Promoter bashing
Promoter bashing is a technique used in molecular biology to identify how certain regions of a DNA strand, commonly promoters, affect the transcription of downstream genes. Under normal circumstances, proteins bind to the promoter and activate or repress transcription. In a promoter bashing assay, specific point mutations or deletions are made in specific regions of the promoter and the transcription of the gene is then measured. The contribution of a region of the promoter can be observed by the level of transcription. If a mutation or deletion changes the level of transcription, then it is known that that region of the promoter may be a binding site or other regulatory element.[1][2][3]
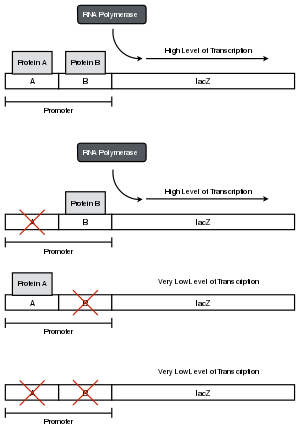
In a laboratory setting, it may not be known that the promoter consists of two regions -- single mutations can be made along the promoter, the promoter can be sequenced, and the levels of reporter assayed to find boundaries for each region.
Promoter bashing is often done with deletions from either the 5' or 3' end of the DNA strand; this assay is easier to perform based on repeated restriction digestion and gel-purifying fragments of specific sizes. It is often easiest to ligate the promoter into the reporter, generate a large amount of the reporter construct using PCR or growth in bacteria, and then perform serial restriction digests on this sample. The ability of upstream promoters can be easily assayed by removing segments from the 5' end, and the same for the 3' end of the strand for downstream promoters.[4]
As the promoter commonly contains binding sequences for proteins affecting transcription, those proteins are also necessary when testing the effects of the promoter. Proteins which associate with the promoter can be identified using an electrophoretic mobility shift assay (EMSA), and the effects of inclusion or exclusion of the proteins with the mutagenized promoters can be assessed in the assay. This allows the use of promoter bashing to not only discover the location on the DNA strand which affects transcription, but also the proteins which affect that strand. The effects of protein interactions with each other as well as the binding sites can also be assayed in this way; candidate proteins must instead be identified by protein/protein interaction assays instead of an EMSA.[5]
Procedure
This is an example procedure for a promoter bashing assay, adapted from Boulin et al.:[6]
- Clone the region of DNA thought to act as a promoter. Cloning is necessary for the assay because it ensures that the promoter is the only factor affecting expression. This step often involves extraction of the DNA from the organism it resides in and PCR amplification.
- Sequence the region. DNA Sequencing is necessary to identify differences in mutated promoters from the wild-type promoter, and to correlate those differences with differences in gene expression. Additionally, it helps with the restriction digest of the region.
- Digest with appropriate restriction endonucleases. The region can be digested to remove elements which are though to not be part of the promoter. Additionally, the reporter gene must be inserted a set distance from the promoter for most promoters. In some methods of promoter bashing, multiple restriction digests are used to systematically remove elements of the promoters—this method ensures that the regions of the promoter removed do not contribute to reporter expression.
- Mutagenize the promoter. Mutating the promoter is necessary if the method of removing part of the promoter with restriction digestion is not used. Many mutated strands can be generated, and the strands sequenced and the activities of the promoters assayed. This is often necessary because one mutation cannot be guaranteed to inactivate a binding site. Non-directed PCR-based mutagenesis can also be used; the parameters of the mutagenic PCR reaction can be adjusted to introduce a reasonable number of mutations. However, the random nature of PCR requires that more strands are assayed downstream of this step.
- Ligate to reporter gene. The promoters to be assayed must be ligated to a reporter gene so that gene expression levels can be measured. The reporter gene must be a sufficient distance from the promoter that the promoter affects it as a wild-type promoter would affect a gene. This can be verified with the positive control (full promoter).
- Transform cells of interest with the various promoter:reporter constructs. The promoter and reporter constructs must be ligated into a plasmid and transformed into cells in which that plasmid can be expressed to measure the activity of each promoter sequence. Proteins which affect the promoter must also be added to those cells — often those proteins are placed on the same or different plasmid under the regulation of a constitutively active promoter.
- Measure reporter-gene transcription rates. The gene products are assayed and the rates of reporter transcription are measured.
From the data received from assaying the different promoters, the effects of various parts of the promoter can be ascertained. However, it is possible that there may not be enough data present and the assay must be re-run with a different promoter region and/or different mutations.
References
- Kamvysselis, M. (2003). Computational molecular genomics: genes, regulation, evolution. (Doctoral Dissertation). Retrieved from http://web.mit.edu/manoli/www/thesis/Intro.html
- Chalfie, M., & Kain, S. (2005) Methods of Biological Analyses, Green Fluorescent Protein: Properties, Applications, and Protocols. Wiley.
- Matsukura, S., Stellato, C., Plitt, J. R., Bickel, C., Miura, K., Georas, S. N., Casolaro, V., Schleimer, R. P. (1999). "Activation of Eotaxin Gene Transcription by NF-κB and STAT6 in Human Airway Epithelial Cells". J Immunol 163:6876-6883. PMID 10586089.
- Engstrom, E. M., Izhaki, A., Bowman, J. L. (2004). "Promoter bashing, microRNAs, and Knox genes. New insights, regulators, and targets-of-regulation in the establishment of lateral organ polarity in Arabidopsis." Plant Phisiol 135(2): 685-94. doi: 10.1104/pp.104.040394. PMID 15208415. PMC 514105.
- Guo, J. Y., Xu, J., Mao, D. Q., Fu, L. L., Gu, J. R., Zhu, J. D. (2002). "The promoter analysis of the human C17orf25 gene, a novel chromosome 17p13.3 gene". Cell Research 12:339-352. doi:10.1038/sj.cr.7290136.
- Boulin, T. et al. Reporter gene fusions (April 5, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi 10.1895/wormbook.1.106.1, http://www.wormbook.org.