PFKFB3
PFKFB3 is a gene that encodes the 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 enzyme in humans.[5][6][7] It is one of 4 tissue-specific PFKFB isoenzymes identified currently (PFKFB1-4).[8]

Gene
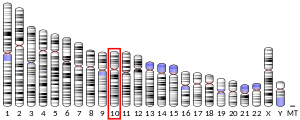
The PFKFB3 gene is mapped to single locus on chromosome 10 (10p15-p14).[5][6] It spans a region of 32.5kb with an open reading frame that is 5,675bp long. It is estimated to consist of 19 exons, of which 15 are regularly expressed.[8] Alternative splicing of the variable, COOH-terminal domain has been observed, leading to 6 different isoforms termed UBI2K1 to UBI2K6 in humans.[9] Different nomenclature also recognizes two broad categories of PFKFB3 isoforms, termed ‘inducible’ and ‘ubiquitous’.[10] The inducible protein isoform, iPFK2, is named as such because its expression has been shown to be induced by hypoxic conditions.
The PFKFB3 promoter is predicted to contain multiple binding sites, including Sp-1 and AP-2 binding sites. It also contains motifs for the binding of E-box, nuclear factor-1 (NF-1), and progesterone response element. Expression of the promoter is shown to be induce by phorbol esters and cyclic-AMP-dependent protein kinase signaling.[10]
Structure
The four PFKFB isoforms share high (85%) ‘2-Kase/2-Pase core’ sequence homology, but have different properties based on variable N- and C- terminal regulatory domains and variation in residues surrounding the active sites.[11] The PFKFB3 inducible isoform has higher ‘2-Kase’ (kinase) activity than other isoforms, due to phosphorylation of Ser-460 by PKA or AMP-dependent protein kinase.[11] The high ‘2-Kase’ activity of PFKFB3 is also due to the lack of a specific Ser that is phosphorylated in the other PFKFB isoforms to decrease kinase activity.[12]
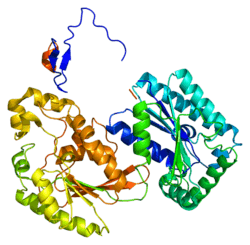
The primary protein encoded by PFKFB3, iPFK2, consists of 590 amino acids. It has a predicted molecular weight of 66.9 kDa and an isoelectric point of 8.64.[8] The crystal structure was determined in 2006:[11]
- Researcher found that iPFK2 has a beta-hairpin N-terminal structure that secures the binding of fructose-6-phosphate to the active site via interaction with the protein's ‘2-Pase’ domain. There are two active pockets within iPFK2 for fructose-2,6-bisphosphatase and 6-phosphofructo-2-kinase which are structurally different. The F-2,6-BP active site structurally open, while the active pocket of 6-phosphofructo-2-kinase is more rigid. This rigidity permits the independent binding of F-6-P and ATP with increased affinity than other isoforms.
Function
iPFK2 converts fructose-6-phosphate to fructose-2,6-bisP (F2,6BP). F2,6BP is a ‘potent’ allosteric activator of 6-phosphofructokinase-1 (PFK-1), stimulating glycolysis. Click to see image of PFFKB3 function.
Role in neuronal excitotoxicity
In neurons, glucose metabolism via glycolysis is usually low when compared to astrocytes. According Astrocyte-to-Neuron Lactate Shuttle Hypothesis, glucose uptake by the brain parenchyma occurs predominantly into astrocytes which subsequently release lactate for the use of neurons.[13] In neurons, glucose is mainly metabolized through the pentose–phosphate pathway (PPP), which is required for NADPH(H+) regeneration and maintenance of neuronal redox status. This neuronal metabolic switch is dictated by the PFKFB3 activity. In neurons, PFKFB3 protein abundance is negligible due to the continuous proteasomal degradation of the enzyme.[14] However, overexcitation of N-methyl-D-aspartate subtype of glutamate receptors (NMDAR), known as excitotoxicity, stabilizes PFKFB3 protein in neurons, resulting in a redirection of glucose flux from PPP to glycolysis, followed by low NADPH(H+) availability for proper GSH regeneration; this ultimately leads to oxidative stress and neuronal death. Silencing of PFKFB3 with small interfering RNA in neurons in vitro prevents the increase in ROS and apoptotic death induced by excitotoxic stimulus.[15] Pharmacological inhibition of PFKFB3 in vitro also protects neurons from apoptosis induced by NMDAR overexcitation as well as from amyloid-ß peptide-induced neurotoxicity. When used in vivo in a mouse model of ischaemic stroke, PFKFB3 inhibitor alleviates motor discoordination and brain infarct injury [16]
Cancer Connections
Warburg Effect
The Warburg effect, proposed by Otto Warbug in 1956,[17] describes the upregulation of glycolysis in most cancer cells, even in the presence of oxygen. The high rate of glycolysis is accompanied by increased lactic acid fermentation, providing additional nutrients for cancer cell growth and tumorigenesis.
PFKFB3 is associated with the Warburg effect because its activity increases the rate of glycolysis. PFKFB3 has been found to be upregulated in numerous cancers, including colon, breast, ovarian, and thyroid.[18] Reduced methylation of PFKFB3 is also found in some cancers, triggering the shift to the glycolytic pathway that supports cancerous growth.[19]
Hypoxia Signaling Pathway
PFKFB3 expression is induced by hypoxia.[20] The promoter of PFKFB3 contains binding sites, called hypoxia response elements (HREs), that recruit the binding of hypoxia-inducible factor-1 (HIF-1).[21]
Hypoxia signaling via HIF-1α stabilization upregulates the transcription of genes that permit survival in low oxygen conditions. These genes include glycolysis enzymes, like PFKFB3, that permit ATP synthesis without oxygen, and vascular endothelial growth factor (VEGF), which promotes angiogenesis.
Cell Cycle & Apoptosis
It was more recently discovered that PFKFB3 promotes cell cycle progression (cell proliferation) and suppresses apoptosis by regulating cyclin-dependent kinase 1 (Cdk-1). PFKFB3's synthesis of F2,6BP in the nucleus was found to regulate Cdk-1, whereas cytosolic PFKFB3 activates PFK-1. Nuclear PFKFB3 activates Cdk1 to phosphorylate the Thr-187 site of p27, causing decreased levels of p27.[22][23] (See summary figure). Reduced p27 causes protection against apoptosis and progression of cells through the G1/S phase checkpoint These findings established a significant link between PFKFB3 cancer cell survival and proliferation.
Circadian Clock
Circadian clocks dysregulation is associated with many types of cancer.[24] PFKFB3 expression exhibits circadian rhythmicity that is different between cancerous and non-cancerous cells.[25] It was specifically found that the circadian-driven transcription factor ‘CLOCK’ binds to the PFKFB3 promoter at a genuine ‘E-box’ site to increase transcription in cancer cells.
- Inhibition of PFKFB3 using 3PO was successful in reducing cancer growth and increasing apoptosis, but only at certain time points within the circadian cycle. This finding highlights the need for time-based PFKFB3 inhibition in cancer treatment. The role of PFKFB3 inhibition in this process should now be considered taking recent information into account that 3PO was shown not to be a PFKFB3 inhibitor (3PO was inactive in a kinase PFKFB3 inhibition assay (IC50 > 100 μM)) [26] (see the corresponding discussion in § Small molecule inhibitors of PFKFB3)
Additional Cancer Connections
- PFKFB3 is activated by progestins in breast cancer cells[27]
- PFKFB3 promotes angiogenesis
- Silencing of PFKFB3 impairs angiogenesis. PFKFB3-driven glycolysis overrules the pro-stalk activity of Notch. PFKFB3 regulates tip and stalk cell behavior and compartmentalizes with F-actin.[28]
Anti-cancer Therapeutic Strategy
Inhibition of PFKFB3 is being analyzed as a potential anti-cancer therapy. The most notable example is clinical trial by Advanced Cancer Therapeutics (ACT) with PFK158, an improved version of 3PO, a PFKFB3 inhibitor. It appears, however, that further development has been discontinued following disappointing Phase I results (see also the discussion of ACT compounds in § Small molecule inhibitors of PFKFB3).
Small molecule inhibitors of PFKFB3
Several small-molecule inhibitors of PFKFB3 are currently in development.
For a long time a small molecule 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) was believed to be an inhibitor of PFKFB3 and used as PFKFB3 inhibitor in many scientific publications. 3PO decreases glucose uptake and increases autophagy.[29] Research is currently exploring various 3PO derivatives (i.e. PFKF15)[30] in an effort to increase their efficacy as anti-cancer therapies, but the data on 3PO derivatives being actually PFKFB3 inhibitors are also unavailable.
Recent research of one of the leading pharmaceuticals companies AstraZeneca and CRT Discovery Laboratories of world's largest independent cancer research charity Cancer Research UK showed that 3PO was inactive in a kinase PFKFB3 inhibition assay (IC50 > 100 μM).[26] The crystal structures of 3PO, as well as its analogues PFK15 and PFK158, with the PFKFB3 enzyme are also not available. The findings of AstraZeneca and Cancer Research UK regarding to 3PO remain unchallenged neither by 3PO developers since April 7, 2015.
The efficacy of two known PFKFB3 inhibitors, namely AZ67 (from AstraZeneca and CRT Discovery Laboratories [26]), and PFK158, an improved but structurally close derivative of 3PO, were recently investigated for their ability to reduce F2,6BP production in A549 cells. Both compounds (AZ67 and PFK158) were able to reduce the cellular levels of F2,6BP in a dose-dependent manner, with IC50 of 0.51 μM and 5.90 μM, respectively. To see if the reduction of cellular F2,6BP levels was a result of direct PFKFB3 inhibition, both compounds were tried in the enzymatic cell-free assay. The study revealed that AZ67 inhibited the enzymatic activity of PFKFB3 with an IC50 of 0.018 μM, a value that is in accordance with previously published results. However, PFK158 had no effect on PFKFB3 enzymatic activity at any of the concentrations tested (up to 100 μM). Accordingly, although PFK158 is able to decrease F2,6BP and glycolytic flux, the experiments show that these effects are not due to PFKFB3 enzymatic inhibition.[16]
Together, these findings put into question the range of scientific research and publications where 3PO and its derivatives (such as PFKF158) was used as a PFKFB3 inhibitor.
In 2018 Kancera reported development and characterization of KAN0438241 (and its pro-drug KAN0438757) as a potent and highly selective PFKFB3 inhibitor and a radiosensitizer.[31]
Other pathways involving PFKFB3
Autophagy
Enhanced activity of PFKFB3 accelerates ROS production as an end product of glycolysis, and thus increases autophagy. Likewise, inhibition of PFKFB3 has been found to induce autophagy.[32][33] See summary image.
Autophagy can prolong cellular survival during low energy conditions. This finding was discovered in relation to rheumatoid arthritis.[34] It was found that RA T cell fail to upregulate autophagy, and knockout experiments placed PFKFB3 as an upstream regulator of this process.
Insulin Signaling Pathway
PFKFB3 was identified in a kinome screen as a regulator of insulin/IGF-1. Suppression of PFKFB3 was found to decrease insulin-stimulated glucose uptake, GLUT4 translocation, and Akt signaling in 3T3-L1 adipocytes. Overexpression caused the insulin-dependent phosphorylation of Akt and Akt substrates.[35]
PFKFB3 expression increases in fat tissues during adipogenesis, but prolonged insulin exposure has been shown to decrease the expression of PFKFB3. This is thought to occur due to a negative feedback mechanism involving insulin.[36]
p38/MK2 Stress Sigaling Pathway
p38 MAPK have been found to increase PFKFB3 activity through (1) the transcriptional activation of PFKFB3 in response to stress stimuli and (2) the post-translational phosphorylation of iPFK2 at Ser-461.[37][38]
See summary figure.[38]
References
- GRCh38: Ensembl release 89: ENSG00000170525 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000026773 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Nicholl J, Hamilton JA, Sutherland GR, Sutherland RL, Watts CK (April 1997). "The third human isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3) map position 10p14-p15". Chromosome Research. 5 (2): 150. doi:10.1023/A:1018482511456. PMID 9146922.
- Manzano A, Rosa JL, Ventura F, Pérez JX, Nadal M, Estivill X, et al. (Mar 1999). "Molecular cloning, expression, and chromosomal localization of a ubiquitously expressed human 6-phosphofructo-2-kinase/ fructose-2, 6-bisphosphatase gene (PFKFB3)". Cytogenetics and Cell Genetics. 83 (3–4): 214–7. doi:10.1159/000015181. PMID 10072580.
- "Entrez Gene: PFKFB3 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3".
- Mahlknecht U, Chesney J, Hoelzer D, Bucala R (October 2003). "Cloning and chromosomal characterization of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 gene (PFKFB3, iPFK2)". International Journal of Oncology. 23 (4): 883–91. doi:10.3892/ijo.23.4.883. PMID 12963966.
- Kessler R, Eschrich K (March 2001). "Splice isoforms of ubiquitous 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in human brain". Brain Research. Molecular Brain Research. 87 (2): 190–5. doi:10.1016/s0169-328x(01)00014-6. PMID 11245921.
- Navarro-Sabaté A, Manzano A, Riera L, Rosa JL, Ventura F, Bartrons R (February 2001). "The human ubiquitous 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene (PFKFB3): promoter characterization and genomic structure". Gene. 264 (1): 131–8. doi:10.1016/S0378-1119(00)00591-6. PMID 11245987.
- Kim SG, Manes NP, El-Maghrabi MR, Lee YH (February 2006). "Crystal structure of the hypoxia-inducible form of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3): a possible new target for cancer therapy". The Journal of Biological Chemistry. 281 (5): 2939–44. doi:10.1074/jbc.M511019200. PMID 16316985.
- Sakakibara R, Kato M, Okamura N, Nakagawa T, Komada Y, Tominaga N, et al. (July 1997). "Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase". Journal of Biochemistry. 122 (1): 122–8. doi:10.1093/oxfordjournals.jbchem.a021719. PMID 9276680.
- Magistretti PJ, Sorg O, Yu N, Martin JL, Pellerin L (1993). "Neurotransmitters regulate energy metabolism in astrocytes: implications for the metabolic trafficking between neural cells". Dev Neurosci. 15 (3–51): 306–12. doi:10.1159/000111349. PMID 7805583.
- Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP (June 2009). "The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1". Nat Cell Biol. 11 (6): 747–52. doi:10.1038/ncb1881. PMID 19448625.
- Rodriguez-Rodriguez P, Fernandez E, Almeida A, Bolaños JP (October 2012). "Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration". Cell Death Differ. 19 (10): 1582–9. doi:10.1038/cdd.2012.33. PMC 3438489. PMID 22421967.
- Burmistrova O, Olias-Arjona A, Lapresa R, Jimenez-Blasco D, Eremeeva T, Shishov D, et al. (August 2019). "Targeting PFKFB3 alleviates cerebral ischemia-reperfusion injury in mice". Scientific Reports. 9 (1): 11670. doi:10.1038/s41598-019-48196-z. PMC 6691133. PMID 31406177.

- Warburg O (February 1956). "On the origin of cancer cells". Science. 123 (3191): 309–14. doi:10.1126/science.123.3191.309. PMID 13298683.
- Atsumi T, Chesney J, Metz C, Leng L, Donnelly S, Makita Z, et al. (October 2002). "High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers". Cancer Research. 62 (20): 5881–7. PMID 12384552.
- Yamamoto T, Takano N, Ishiwata K, Ohmura M, Nagahata Y, Matsuura T, et al. (March 2014). "Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway". Nature Communications. 5: 3480. doi:10.1038/ncomms4480. PMC 3959213. PMID 24633012.
- Minchenko A, Leshchinsky I, Opentanova I, Sang N, Srinivas V, Armstead V, Caro J (February 2002). "Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect". The Journal of Biological Chemistry. 277 (8): 6183–7. doi:10.1074/jbc.M110978200. PMC 4518871. PMID 11744734.
- Obach M, Navarro-Sabaté A, Caro J, Kong X, Duran J, Gómez M, et al. (December 2004). "6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia". The Journal of Biological Chemistry. 279 (51): 53562–70. doi:10.1074/jbc.M406096200. PMID 15466858.
- Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, et al. (September 2009). "Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases". The Journal of Biological Chemistry. 284 (36): 24223–32. doi:10.1074/jbc.M109.016816. PMC 2782016. PMID 19473963.
- Yalcin A, Clem BF, Imbert-Fernandez Y, Ozcan SC, Peker S, O'Neal J, et al. (July 2014). "6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27". Cell Death & Disease. 5 (7): e1337. doi:10.1038/cddis.2014.292. PMC 4123086. PMID 25032860.
- Savvidis C, Koutsilieris M (December 2012). "Circadian rhythm disruption in cancer biology". Molecular Medicine. 18 (1): 1249–60. doi:10.2119/molmed.2012.00077. PMC 3521792. PMID 22811066.
- Chen L, Zhao J, Tang Q, Li H, Zhang C, Yu R, et al. (April 2016). "PFKFB3 Control of Cancer Growth by Responding to Circadian Clock Outputs". Scientific Reports. 6: 24324. doi:10.1038/srep24324. PMC 4832144. PMID 27079271.
- Boyd S, Brookfield JL, Critchlow SE, Cumming IA, Curtis NJ, Debreczeni J, et al. (April 2015). "Structure-Based Design of Potent and Selective Inhibitors of the Metabolic Kinase PFKFB3". Journal of Medicinal Chemistry. 58 (8): 3611–25. doi:10.1021/acs.jmedchem.5b00352. PMID 25849762.
- Novellasdemunt L, Obach M, Millán-Ariño L, Manzano A, Ventura F, Rosa JL, et al. (March 2012). "Progestins activate 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) in breast cancer cells". The Biochemical Journal. 442 (2): 345–56. doi:10.1042/BJ20111418. hdl:10261/87967. PMID 22115192.
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. (August 2013). "Role of PFKFB3-driven glycolysis in vessel sprouting". Cell. 154 (3): 651–63. doi:10.1016/j.cell.2013.06.037. PMID 23911327.
- Klarer AC, O'Neal J, Imbert-Fernandez Y, Clem A, Ellis SR, Clark J, et al. (January 2014). "Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism". Cancer & Metabolism. 2 (1): 2. doi:10.1186/2049-3002-2-2. PMC 3913946. PMID 24451478.
- Clem BF, O'Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA, et al. (August 2013). "Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer". Molecular Cancer Therapeutics. 12 (8): 1461–70. doi:10.1158/1535-7163.MCT-13-0097. PMC 3742633. PMID 23674815.
- Gustafsson NM, Färnegårdh K, Bonagas N, Ninou AH, Groth P, Wiita E, et al. (September 2018). "Targeting PFKFB3 radiosensitizes cancer cells and suppresses homologous recombination". Nature Communications. 9 (1): 3872. doi:10.1038/s41467-018-06287-x. PMC 6155239. PMID 30250201.
- Klarer AC, O'Neal J, Imbert-Fernandez Y, Clem A, Ellis SR, Clark J, et al. (January 2014). "Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism". Cancer & Metabolism. 2 (1): 2. doi:10.1186/2049-3002-2-2. PMC 3913946. PMID 24451478.
- Yang Z, Goronzy JJ, Weyand CM (February 2014). "The glycolytic enzyme PFKFB3/phosphofructokinase regulates autophagy". Autophagy. 10 (2): 382–3. doi:10.4161/auto.27345. PMC 5079104. PMID 24351650.
- Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM (September 2013). "Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells". The Journal of Experimental Medicine. 210 (10): 2119–34. doi:10.1084/jem.20130252. PMC 3782046. PMID 24043759.
- Trefely S, Khoo PS, Krycer JR, Chaudhuri R, Fazakerley DJ, Parker BL, et al. (October 2015). "Kinome Screen Identifies PFKFB3 and Glucose Metabolism as Important Regulators of the Insulin/Insulin-like Growth Factor (IGF)-1 Signaling Pathway". The Journal of Biological Chemistry. 290 (43): 25834–46. doi:10.1074/jbc.M115.658815. PMC 4646237. PMID 26342081.
- Atsumi T, Nishio T, Niwa H, Takeuchi J, Bando H, Shimizu C, et al. (December 2005). "Expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase/PFKFB3 isoforms in adipocytes and their potential role in glycolytic regulation". Diabetes. 54 (12): 3349–57. doi:10.2337/diabetes.54.12.3349. PMID 16306349.
- Novellasdemunt L, Bultot L, Manzano A, Ventura F, Rosa JL, Vertommen D, et al. (June 2013). "PFKFB3 activation in cancer cells by the p38/MK2 pathway in response to stress stimuli". The Biochemical Journal. 452 (3): 531–43. doi:10.1042/bj20121886. PMID 23548149.
- Bolaños JP (June 2013). "Adapting glycolysis to cancer cell proliferation: the MAPK pathway focuses on PFKFB3". The Biochemical Journal. 452 (3): e7-9. doi:10.1042/bj20130560. PMID 23725459.
Further reading
- Sakai A, Kato M, Fukasawa M, Ishiguro M, Furuya E, Sakakibara R (March 1996). "Cloning of cDNA encoding for a novel isozyme of fructose 6-phosphate, 2-kinase/fructose 2,6-bisphosphatase from human placenta". Journal of Biochemistry. 119 (3): 506–11. doi:10.1093/oxfordjournals.jbchem.a021270. PMID 8830046.
- Hamilton JA, Callaghan MJ, Sutherland RL, Watts CK (April 1997). "Identification of PRG1, a novel progestin-responsive gene with sequence homology to 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase". Molecular Endocrinology. 11 (4): 490–502. doi:10.1210/me.11.4.490. PMID 9092801.
- Sakakibara R, Kato M, Okamura N, Nakagawa T, Komada Y, Tominaga N, et al. (July 1997). "Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase". Journal of Biochemistry. 122 (1): 122–8. doi:10.1093/oxfordjournals.jbchem.a021719. PMID 9276680.
- Scanlan MJ, Gordan JD, Williamson B, Stockert E, Bander NH, Jongeneel V, et al. (November 1999). "Antigens recognized by autologous antibody in patients with renal-cell carcinoma". International Journal of Cancer. 83 (4): 456–64. doi:10.1002/(SICI)1097-0215(19991112)83:4<456::AID-IJC4>3.0.CO;2-5. PMID 10508479.
- Fukasawa M, Takayama E, Shinomiya N, Okumura A, Rokutanda M, Yamamoto N, Sakakibara R (January 2000). "Identification of the promoter region of human placental 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene". Biochemical and Biophysical Research Communications. 267 (3): 703–8. doi:10.1006/bbrc.1999.2022. PMID 10673355.
- Kessler R, Eschrich K (March 2001). "Splice isoforms of ubiquitous 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in human brain". Brain Research. Molecular Brain Research. 87 (2): 190–5. doi:10.1016/S0169-328X(01)00014-6. PMID 11245921.
- Riera L, Obach M, Navarro-Sabaté A, Duran J, Perales JC, Viñals F, et al. (August 2003). "Regulation of ubiquitous 6-phosphofructo-2-kinase by the ubiquitin-proteasome proteolytic pathway during myogenic C2C12 cell differentiation". FEBS Letters. 550 (1–3): 23–9. doi:10.1016/S0014-5793(03)00808-1. PMID 12935880.
- Obach M, Navarro-Sabaté A, Caro J, Kong X, Duran J, Gómez M, et al. (December 2004). "6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia". The Journal of Biological Chemistry. 279 (51): 53562–70. doi:10.1074/jbc.M406096200. PMID 15466858.
- Manes NP, El-Maghrabi MR (June 2005). "The kinase activity of human brain 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase is regulated via inhibition by phosphoenolpyruvate". Archives of Biochemistry and Biophysics. 438 (2): 125–36. doi:10.1016/j.abb.2005.04.011. PMID 15896703.
- Minchenko OH, Ogura T, Opentanova IL, Minchenko DO, Esumi H (December 2005). "Splice isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4: expression and hypoxic regulation". Molecular and Cellular Biochemistry. 280 (1–2): 227–34. doi:10.1007/s11010-005-8009-6. PMID 16311927.
PDB gallery | |
|---|---|
|
{kind=link}