Mullerian anomalies
Mullerian duct anomalies are those structural anomalies caused by errors in müllerian-duct development during embryonic morphogenesis. Factors that precipitate include genetics, and maternal exposure to teratogens.[1][2]
Genetic causes of müllerian duct anomalies are complicated and uncommon. Inheritance patterns can be autosomal dominant, autosomal recessive, and X-linked disorders. Müllerian anomalies can be part of a multiple malformation syndrome.[1][3]
Mullerian anomalies occur as a congenital malformation of the mullerian ducts during embryogenesis. The mullerian ducts are also referred to as paramesonephric ducts, referring to ducts next to (para) the mesonephric (Wolffian) duct during foetal development. Paramesonephric ducts are paired ducts derived from the embryo, and for females develop into the uterus, uterine tubes, cervix and upper two third of the vagina.[4] Embryogenesis of the mullerian ducts play important roles in ensuring normal development of the female reproductive tract. However, when defects in each of the three phases of embryogenesis occur, it results in specific structural malformations which are distinguished according to anatomy into seven classes based on the American Society for Reproductive Medicine (ASRM) classification system.[5] Class I and II anomalies result from underdevelopment of the two separate primitive uterine, vaginal and cervical pockets due to an arrest of stage one of organogenesis, resulting in underdevelopment of both left and right primitive uterus (class I) or underdevelopment of one of the primitive uterus (class II). Class III and IV anomalies result from failure of midline fusion of the two separate primitive pockets due to an arrest of stage two of organogenesis. Class V and VI anomalies result from failure of degeneration of the midline due to an arrest at stage three of organogenesis. Class VII anomalies are malformations caused by Diethylstilbestrol (DES).[6]
Vaginal agenesis (Mayer–Rokitansky–Kuster–Hauser syndrome) and Class I anomalies
Mayer–Rokitansky–Kuster–Hauser (MRKH) syndrome is a class I developmental disorder of the mullerian ducts where the vagina and uterus are underdeveloped or absent. Females with MRHK syndrome have normal chromosome pattern of 46,XX karyotype, with normal functioning ovaries and secondary sex characteristics.[7] Females with MRKH are unable to carry a pregnancy due to a malformed uterus, but can have children via assisted reproduction. MRKH syndrome type 1 results when only reproductive organs such as vagina are affected (vaginal agenesis) and type 2 results when abnormalities develop in other parts of the body such as abnormal kidney formation (unilateral renal agenesis).[8]
Causes
MRHK syndrome occurs from an arrest in the embryonic development in the first phase of organogenesis, resulting in underdevelopment of one or both the left and right primitive uterus and vagina (agenesis). Specifically, an arrest in the development of the paramesonephric ducts at week seven of gestation is linked to MRHK syndrome.[7] During embryological development of the first phase, two separate uterine, cervical and vaginal pockets develop, following which a transverse septum forms across the caudal aspect in the upper two thirds of the vagina, which will dissolve when the lower one third of the vagina (developed from the urogenital sinus) fuses with the upper two thirds. An arrest at this stage means midline fusion of pockets do not occur and subsequently are unable to develop into a whole uterus, cervix and vagina.[9]
Changes in the sequences of DNA bases due to mutations in WNT3, HNF1b and LHX1 are decreased in people with MRKH.[10] Mice with mutant alleles for Wnt4, Wnt5a, Wnt7a and Wnt9b display varying extents of mullerian duct hypoplasia indicating these genes may cause MRKH-like phenotypes in humans.[11] A commonly identified copy number variants (CNVs) deletion of 17q12 is present in both type 1 and type 2 MRKH patients. The deletion of 17q12 results in a loss of 2 specific genes, HNFB and LHX1 at position q12 on chromosome 17 which are linked to mullerian anomalies. Most 17q12 deletions result from genetic mutations in people with no known history of MRHK syndrome in their family.[10]
Clinical presentations and diagnosis
Patients who have not reached puberty are asymptomatic and diagnosis is difficult at this stage as the vagina and uterus are not fully developed. Estrogenization during puberty will increase the size of the uterus and allow for accurate evaluation. Symptomatic patients will present with pain at the uterus area due to infections or abnormal vaginal bleeding with cyclical pelvic pain.[12]
Clinical presentations of Septate uterus
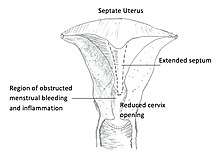
Diagnosis of septate uterus is based on ultrasound findings of two endometrial cavities and a smooth contouring of the fundus. The septum separating both endometrial cavities is thin and may descend into the cervix of the vagina.[13] An over extended septum can cause the cervix to be obstructed, allowing pathogens to infect the region resulting in pelvic pain due to inflammation of the cervix. The Mullerian duct can be partially obstructed or fully obstructed. In the case where the Mullerian duct is partially obstructed, a reduced cervix opening obstructs menstrual bleeding flow, causing prolonged menstrual bleeding (hypomenorrhea). When there is complete obstruction, patients will present with absence of menstruation (amenorrhea).[14] For patients where the septum extends longitudinally, bleeding will persist when a tampon is used as there are two vaginal openings, and dyspareunia (pain during intercourse) is common.[15]
Magnetic Resonance Imaging (MRI) is useful in detecting obstruction of the endometrium due to hematometra, which appears as cavitated uterine buds on images, and are unable to be detected by ultrasound. MRI provides three-dimensional information of both internal and external contours and can differentiate septate from bicornuate uterus and other complex anomalies.[16]
Development defects in other tissues
Malformation of the mullerian ducts in foetuses can result in exhibition of extragenital anomalies such as urological anomalies that includes unilateral renal agenesis, horseshoe kidneys or malformation of collecting ducts. Skeletal malformations which include congenital dislocation of the hip, malformations of the arms, foot, ribs, hemivertebrae in the lumbar spine and cervical spina bifida are associated with mullerian anomalies.[17] Mutations of homeobox genes HOXA10, HOXA11 and HOXA13 in uterus malformations are also responsible for renal and skeletal developmental anomalies.[18] However, the mechanism of action of these genes has not been established.
Non-surgical treatment
The Frank and Ingram procedure is a common non-operative procedure used to increase function of the vaginal via dilators. The method uses graduated dilators to progressively invaginate the mucosa to dilate the opening, increasing depth and functionality of the vaginal over time. The Ingram modification involves using a bicycle seat positioned between the legs allowing direct contact with the perineum creating pressure on the vagina. Thus, by applying pressure to the mucosa, a neovagina forms. It takes between four months up to several years for complete successful treatment.[19]
Surgical treatment
The McIndoe procedure uses a split-thickness skin graft from the patient where it is placed over an obturator and sewn at the ends to from a tube with one closed end. A transverse incision at the vaginal dimple and a small cavity is made at the level of the peritoneum by the surgeon. The skin graft and obturator are inserted into the vagina vault and secured to the labia minora. Synthetic skin grafts are also an alternative, eliminating the need for skin grafts from patients.[20] The use of dilators post operation for three to six months is required to prevent contraction of the vagina. Complications include skin graft failure due to the formation of a hematoma beneath the graft, postoperative hematoma that prevents the graft from receiving adequate nourishment, rectal perforation and fistula formation. Patients with prior history of vaginal or perineal surgery have higher complication rates.[9]
The Sigmoid vaginaplasty procedure uses a segment of the patient’s sigmoid colon where one end is pulled down to form a neovagina while the other end is sealed forming a blind pouch. Complications include narrowing of the vaginal (stenosis) and weakening of pelvic floor muscles and ligaments which are unable to support the uterus (uterine prolapse). As the use of dilators are not required for sigmoid vaginoplasty, this treatment is favoured over the McIndoe procedure.[21]
Diethylstilbestrol and class VII anomalies
Diethylstilbestrol (DES) was a synthetic oestrogen supplement introduced in 1938 to decrease miscarriage in the first trimester by enhancing the oestrogen dependent follicular phase and implantation of blastocysts. DES is a known teratogen, by crossing the placenta DES disrupts organogenesis by disorganising uterine muscle layers causing maldevelopment of uterus and uterine tube junctions. This prevents normal columnar ciliated cell formation of the vaginal epithelium and reabsorption of vaginal glands.[22] When absorbed, DES is broken down to produce a transient quinone-like reactive intermediate that alters normal gene function of HOX and WNT, affecting differentiation of mullerian ducts. In utero DES exposure has additionally been linked to epigenetic changes responsible for uterine anomalies such as dysregulation of the homeobox gene HOXA10 by hypermethylation of HOXA10, altering long term expression of genes which controls uterine organogenesis.[23]
DES is also an endocrine disrupting compound (EDC) which alters normal hormone responses required for reproductive tract development in foetuses. A dose–response association for DES has not been establish but an association with the time of exposure in utero suggest exposure to DES at a certain embryological stage leads to increase susceptibility to mullerian anomalies.[22]
Female foetuses exposed to DES in utero (DES daughters) have abnormalities in development in three areas of the mullerian duct, namely of their uterus, cervix and vagina. DES uterine anomalies include hypoplastic uterus (small uterus), T-shaped uterine cavity and constrictions of the endometrial cavity.[24] DES uterine anomalies vary in extent in different races, with foetuses of African American females being more prone to fibroids development during organogenesis.[25] DES cervical and vaginal anomalies include hypoplasia, collar and hood malformation of the vagina and cervix and is seen in 20% of women exposed to DES.[24]
Epidemiology
The prevalence of vaginal agenesis or class I uterine anomalies is 1:5000 female live births globally. The most prevalent form of vaginal agenesis is Mayer–Rokitansky–Kuster–Hauser (MRKH) syndrome and results in congenital aplasia or hypoplasia of mullerian derived structures.[7] MRKH syndrome account for 5% to 10% of all mullerian anomalies. While septate uterus or class II uterine anomalies account for 3% to 7% of all mullerian anomalies. The prevalence of mullerian anomalies also differs within the female population, occurring in 5.5% of the general population, 8% in sterile females and 13.3% in females with a history of miscarriage.[26]
Pathogenesis
The human female reproductive system consists of the gonads, external genitalia and the Mullerian duct system.[27] Initially in the embryo, both the Wolffian (mesonephric) and Mullerian (paramesonephric) ducts are present, where development of the Wolffian ducts give rise to the male reproductive tract and development of the Mullerian ducts give rise to the female reproductive tract.[27][28] These ducts are identical until approximately week 6 of embryonic development. In males, the sex-determining region Y (SRY) gene on the Y chromosome suppresses Mullerian duct development, by initiating the production of anti-Mullerian hormone by the Sertoli cells of the testis. The Mullerian ducts only develop in the absence of anti-Mullerian hormone, where the Wolffian ducts regress.
Development of the female reproductive tract begins at approximately week 8 of embryonic development, and development of the Mullerian duct system is typically complete by the end of the first trimester.[28][29] The Mullerian ducts develop to give rise to the fallopian tubes, uterus, cervix and upper two-thirds of the vagina. The ovaries are not part of the Mullerian system and arise from primordial germ cells, which develop at the gonadal ridge.
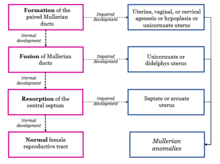
The formation of the female reproductive tract via the Mullerian ducts has 3 distinct stages. An array of Mullerian anomalies can occur if any of these processes are arrested or impaired.[28][29][30]
The first stage of Mullerian duct development is organogenesis, where both Mullerian ducts are formed.[29] If the formation of the Mullerian ducts is impaired or does not occur, this can give rise to uterine, cervical and/or vaginal hypoplasia or agenesis.[28] Mullerian agenesis, also known as the Mayer–Rokitansky–Kuster–Hauser (MRKH) syndrome, results in the congenital absence of the vagina or uterus.[31] Women with MRKH syndrome commonly present with primary amenorrhea, where menstruation does not occur by the age of 16. In the first stage of development, it is also possible for only one Mullerian duct to develop, giving rise to a single uterine horn (unicornuate uterus).[28] Unicornuate uteri commonly develop on the right side, although the reason for this preference remains elusive.[27]
The second stage of Mullerian duct development involves the fusion of the inferior portion of the ducts to form the uterus, cervix and upper two-thirds of the vagina.[29]The superior part of the Mullerian ducts do not fuse and form the left and right fallopian tubes. Disruptions to this stage of development can result in didelphys or bicornuate uteri anomalies.[28] In both didelphys and bicornuate uteri, the non-fusion of the Mullerian ducts results in two distinct uterine cavities.
The third and final stage of Mullerian duct development is septal resorption.[28] After the lower Mullerian ducts fuse, a central septum is left behind, and this partition must be eliminated to give rise to a single uterine cavity, cervical canal and vaginal canal. Defects in septal resorption may produce a septate uterus or arcuate uterus, where the septum divides the uterine cavity. More than 50% of women with reported Mullerian anomalies have septate uteri.[27] It is common for other developmental defects to occur in conjunction with Mullerian anomalies, including renal, skeletal, auditory and cardiac abnormalities.[30][31]
Causes
The causes of Mullerian anomalies are not well-understood.[29][30][31][32] The aetiology of this congenital disease may be multifactorial, with genetics, socioeconomic factors and geographic factors playing a role in dysfunctional Mullerian duct development.[29] Mullerian anomalies likely occur early in development, as the congenital disorder often occurs in association with renal and anorectal disorders.[30][31]
Typically, women with Mullerian abnormalities have a normal female karyotype (46, XX). Most incidences of Mullerian anomalies occur sporadically, with instances of familial inheritance patterns being less common.[30] The genetic component of the disease classically follows an autosomal dominant pattern, with variable rates of genotypic expression.
WNT4 signalling
WNT4 is a gene that has a crucial role in embryonic development, particularly to ensure the normal formation of the female reproductive system, the kidneys and several endocrine organs.[33] The Wnt4 gene pathway promotes female sexual differentiation, while suppressing male sexual differentiation. Mice lacking Wnt4 display androgenisation, the presence of Wolffian ducts and absence of Mullerian ducts. This effect is mirrored in humans, where mutations in the WNT4 gene has been observed in MRKH syndrome patients, who display hyperandrogenism. Mutations in WNT4 gene are not always present in individuals with Mullerian anomalies or MRKH syndrome, but the WNT4 gene is the only gene that has been clearly implicated in MRKH.[30]
TP63
TP63 is a tumour protein encoded by the EMX2 gene, which is expressed in uterine and vaginal epithelium.[32] The TP63 protein is required for epithelial differentiation during Mullerian duct development in utero, by promoting the transcription of particular genes. EMX2 mutations result in incomplete Mullerian fusion. Some women with unicornuate uteri exhibit mutant EMX2 and significantly decreased expression of TP63, implicating TP63 in the fusion stage of Mullerian development.[7]
Diethylstilbestrol (DES)
DES is a synthetic non-steroidal estrogen that was used during 1940–1971, to prevent premature births, miscarriage and other pregnancy complications.[34][35] The use of DES was discontinued after it was established that approximately 69% of females who were exposed to DES in utero had uterine abnormalities. [35] DES has been marked as a teratogen as it results in malformation of the embryo. DES is more potent than steroidal estrogen and binds to cytosolic receptors after crossing the placenta.[36] DES is not metabolised as quickly as endogenous estrogen. DES remains bound to cytosolic receptors for a longer period of time. The extended binding time of DES and the subsequent prolonged activation of its cognate receptors has been suggested to disrupt Mullerian development, resulting in uterine abnormalities. Exposure to DES induced multiple uterine abnormalities including constriction bands, hypoplasticity in the uterine cavity and irregular borders. Females exposed to this teratogen in utero presented most commonly with a T-shaped uterus, resulting in increased rates of ectopic pregnancy, spontaneous abortions, and an overall increased risk of adverse pregnancy outcome.[35]
Impact on fertility and pregnancy
The incidence of individuals with Mullerian anomalies is twice as high in the infertile population than in the fertile population.[27] Women who do experience some obstetric complications usually have trouble maintaining full-term pregnancy, rather than issues with conception. Due to improper development of the uterus and fallopian tubes, pregnancies in women with Mullerian anomalies could result in spontaneous abortions, preterm birth, intrauterine growth restriction, perinatal mortality, placental abruption and other malpresentations.[37][38][39]
Advancements in the epidemiology of Mullerian anomalies has resulted in earlier diagnosis and treatment. Uterine obstructions can be surgically repaired or managed to result in successful perinatal outcomes.[38]
Normal ovarian function is not interrupted in females with Mullerian anomalies and women with the anomaly have been able to utilise assisted reproductive technologies and a gestational carrier to increase the chance of successful reproductive outcomes.[40]
Maintaining pregnancy
Physiological changes that occur in conjunction with Mullerian anomalies explain why some women with the disorder experience difficulties maintaining pregnancy. These physiological changes include compromised blood flow to the uterus, low uterine muscle mass and an insufficient cervix.[38][39]
An insufficient flow of blood to the uterus would compromise nutritional supply to the foetus and waste removal from the foetus, and this can explain the heightened occurrence of low foetal birth weight (intrauterine growth restriction) and spontaneous abortions in women with Mullerian anomalies.[38][40] Women with anomalies such as didelphys and bicornuate uteri present with a decreased uterine size and subsequent lower muscle mass.[37][38] A diminished uterine capacity reduces the likelihood of the foetus reaching full-term development due to spatial constraints, explaining the higher rates of preterm births observed in women with Mullerian anomalies.
The degree to which the Mullerian anomaly impairs the reproductive potential of a woman varies between individuals, and is dependent on the type of anomaly and its severity.[27] Women with minor fusion defects such as arcuate uteri and septate uteri tend to have a lower risk of aversive pregnancy outcome, compared to patients with major fusion defects, such as unicornuate uteri, bicornuate uteri and didelphys uteri.[41] Females with severe agenesis and/or hypoplasia, such as in MRKH syndrome, have an increased chance of poor reproductive outcomes without surgical intervention.[31]
Assisted reproductive technologies
Women with Mullerian anomalies often utilise assisted reproductive technologies such as in vitro fertilisation (IVF), intracytoplasmic sperm injection (ICSI) and embryo transfer (ET), and/or a gestational carrier.[42] Compared to individuals with no uterine anomalies, women with Mullerian anomalies exhibit no differences in number of follicles produced, number of oocytes retrieved, or levels of estrogen produced.[43] The normal follicular function, oocyte population and estrogen levels in women with Mullerian anomalies occurs as normal ovarian function is not compromised in the disease.
In patients with uterine anomalies, there may be a chance that the endometrial cavity can be compromised, such that implantation following IVF does not always lead to a successful pregnancy.[42][43] If an individual has a reduced likelihood of effective implantation, a gestational carrier can be appointed to increase the chance of a successful pregnancy.[40][44]
Women that present with unicornuate uteri may have an increased risk of spontaneous abortions, premature labour and preterm delivery, while individuals with unicornuate uteri may be at risk of ectopic pregnancy.[34][38] These risks can be minimised if assisted reproductive technologies are utilised.
Correcting the anomaly prior to commencing assisted reproductive technologies can increase the possibility of reproductive success by increasing the chance of implantation and reducing the likelihood of complications occurring after pregnancy occurs.[38] A greater rate of successful pregnancies are observed in women with septate uteri when the septum is operated on prior to implantation of the embryo.[45]
See also
- Mayer–Rokitansky–Küster–Hauser syndrome
- Vaginal agenesis
References
- Amesse LS (June 2016). "Mullerian Duct Anomalies: Overview, Incidence and Prevalence, Embryology". Medscape. WebMD.
- "Mullerian Anomalies". Penn Medicine. University of Pennsylvania. Retrieved 2018-01-21.
- "What is Vaginal Agenesis?". Urology Care Foundation. Retrieved 2018-01-21.
- Mullen RD, Behringer RR (2014). "Molecular genetics of Müllerian duct formation, regression and differentiation". Sexual Development. 8 (5): 281–96. doi:10.1159/000364935. PMC 4378544. PMID 25033758.
- Chandler TM, Machan LS, Cooperberg PL, Harris AC, Chang SD (December 2009). "Mullerian duct anomalies: from diagnosis to intervention". The British Journal of Radiology. 82 (984): 1034–42. doi:10.1259/bjr/99354802. PMC 3473390. PMID 19433480.
- Robbins JB, Broadwell C, Chow LC, Parry JP, Sadowski EA (January 2015). "Müllerian duct anomalies: embryological development, classification, and MRI assessment". Journal of Magnetic Resonance Imaging. 41 (1): 1–12. doi:10.1002/jmri.24771. PMID 25288098.
- Fontana L, Gentilin B, Fedele L, Gervasini C, Miozzo M (February 2017). "Genetics of Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome". Clinical Genetics. 91 (2): 233–246. doi:10.1111/cge.12883. PMID 27716927.
- Robbins JB, Broadwell C, Chow LC, Parry JP, Sadowski EA (January 2015). "Müllerian duct anomalies: embryological development, classification, and MRI assessment". Journal of Magnetic Resonance Imaging. 41 (1): 1–12. doi:10.1002/jmri.24771. PMID 25288098.
- Aiguo W, Guangren D (2007). "PMID Observer Design of Descriptor Linear Systems". 2007 Chinese Control Conference. IEEE: 161–165. doi:10.1109/chicc.2006.4347343. ISBN 9787811240559.
- Rasmussen M, Vestergaard EM, Graakjaer J, Petkov Y, Bache I, Fagerberg C, Kibaek M, Svaneby D, Petersen OB, Brasch-Andersen C, Sunde L (November 2016). "17q12 deletion and duplication syndrome in Denmark – A clinical cohort of 38 patients and review of the literature". American Journal of Medical Genetics. Part A. 170 (11): 2934–2942. doi:10.1002/ajmg.a.37848. PMID 27409573.
- Williams LS, Demir Eksi D, Shen Y, Lossie AC, Chorich LP, Sullivan ME, Phillips JA, Erman M, Kim HG, Alper OM, Layman LC (July 2017). "Genetic analysis of Mayer–Rokitansky–Kuster–Hauser syndrome in a large cohort of families". Fertility and Sterility. 108 (1): 145–151.e2. doi:10.1016/j.fertnstert.2017.05.017. PMC 5770980. PMID 28600106.
- Colvin CW, Abdullatif H (January 2013). "Anatomy of female puberty: The clinical relevance of developmental changes in the reproductive system". Clinical Anatomy. 26 (1): 115–29. doi:10.1002/ca.22164. PMID 22996962.
- Heinonen PK (March 2006). "Complete septate uterus with longitudinal vaginal septum". Fertility and Sterility. 85 (3): 700–5. doi:10.1016/j.fertnstert.2005.08.039. PMID 16500341.
- Kamio M, Nagata C, Sameshima H, Togami S, Kobayashi H (July 2018). "Obstructed hemivagina and ipsilateral renal anomaly (OHVIRA) syndrome with septic shock: A case report". The Journal of Obstetrics and Gynaecology Research. 44 (7): 1326–1329. doi:10.1111/jog.13656. PMID 29978541.
- de França Neto AH, Nóbrega BV, Clementino Filho J, do Ó TC, de Amorim MM (2014). "Intrapartum diagnosis and treatment of longitudinal vaginal septum". Case Reports in Obstetrics and Gynecology. 2014: 108973. doi:10.1155/2014/108973. PMC 4033546. PMID 24891963.
- Pellerito JS, McCarthy SM, Doyle MB, Glickman MG, DeCherney AH (June 1992). "Diagnosis of uterine anomalies: relative accuracy of MR imaging, endovaginal sonography, and hysterosalpingography". Radiology. 183 (3): 795–800. doi:10.1148/radiology.183.3.1584936. PMID 1584936.
- Bhagavath B, Ellie G, Griffiths KM, Winter T, Alur-Gupta S, Richardson C, Lindheim SR (June 2017). "Uterine Malformations: An Update of Diagnosis, Management, and Outcomes". Obstetrical & Gynecological Survey. 72 (6): 377–392. doi:10.1097/OGX.0000000000000444. PMID 28661551.
- Choussein S, Nasioudis D, Schizas D, Economopoulos KP (June 2017). "Mullerian dysgenesis: a critical review of the literature". Archives of Gynecology and Obstetrics. 295 (6): 1369–1381. doi:10.1007/s00404-017-4372-2. PMID 28434104.
- Jasonni VM, La Marca A, Naldi S, Matonti G, D'Anna R (December 2007). "The management of vaginal agenesis: report of 104 cases". Fertility and Sterility. 88 (6): 1653–6. doi:10.1016/j.fertnstert.2007.01.126. PMID 17481623.
- Reichman DE, Laufer MR (April 2010). "Congenital uterine anomalies affecting reproduction". Best Practice & Research. Clinical Obstetrics & Gynaecology. 24 (2): 193–208. doi:10.1016/j.bpobgyn.2009.09.006. PMID 19897423.
- Theodoridis TD, Pappas PD, Grimbizis GF (February 2019). "Surgical management of congenital uterine anomalies (including indications and surgical techniques)". Best Practice & Research. Clinical Obstetrics & Gynaecology. 59: 66–76. doi:10.1016/j.bpobgyn.2019.02.006. PMID 30910446.
- Reed CE, Fenton SE (June 2013). "Exposure to diethylstilbestrol during sensitive life stages: a legacy of heritable health effects". Birth Defects Research. Part C, Embryo Today. 99 (2): 134–46. doi:10.1002/bdrc.21035. PMC 3817964. PMID 23897597.
- Bromer JG, Wu J, Zhou Y, Taylor HS (July 2009). "Hypermethylation of homeobox A10 by in utero diethylstilbestrol exposure: an epigenetic mechanism for altered developmental programming". Endocrinology. 150 (7): 3376–82. doi:10.1210/en.2009-0071. PMC 2703508. PMID 19299448.
- Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, Cheville AL, Colton T, Hartge P, Hatch EE, Herbst AL, Karlan BY, Kaufman R, Noller KL, Palmer JR, Robboy SJ, Saal RC, Strohsnitter W, Titus-Ernstoff L, Troisi R (October 2011). "Adverse health outcomes in women exposed in utero to diethylstilbestrol". The New England Journal of Medicine. 365 (14): 1304–14. doi:10.1056/NEJMoa1013961. PMID 21991952.
- D'Aloisio AA, Baird DD, DeRoo LA, Sandler DP (March 2012). "Early-life exposures and early-onset uterine leiomyomata in black women in the Sister Study". Environmental Health Perspectives. 120 (3): 406–12. doi:10.1289/ehp.1103620. PMC 3295338. PMID 22049383.
- Chan YY, Jayaprakasan K, Zamora J, Thornton JG, Raine-Fenning N, Coomarasamy A (2011). "The prevalence of congenital uterine anomalies in unselected and high-risk populations: a systematic review". Human Reproduction Update. 17 (6): 761–71. doi:10.1093/humupd/dmr028. PMC 3191936. PMID 21705770.
- Shulman LP (June 2008). "Müllerian anomalies". Clinical Obstetrics and Gynecology. 51 (2): 214–22. doi:10.1097/GRF.0b013e31816feba0. PMID 18463453.
- Robbins JB, Broadwell C, Chow LC, Parry JP, Sadowski EA (January 2015). "Müllerian duct anomalies: embryological development, classification, and MRI assessment". Journal of Magnetic Resonance Imaging. 41 (1): 1–12. doi:10.1002/jmri.24771. PMID 25288098.
- Saravelos SH, Cocksedge KA, Li TC (2008). "Prevalence and diagnosis of congenital uterine anomalies in women with reproductive failure: a critical appraisal". Human Reproduction Update. 14 (5): 415–29. doi:10.1093/humupd/dmn018. PMID 18539641.
- Cheroki C, Krepischi-Santos AC, Szuhai K, Brenner V, Kim CA, Otto PA, Rosenberg C (April 2008). "Genomic imbalances associated with mullerian aplasia". Journal of Medical Genetics. 45 (4): 228–32. doi:10.1136/jmg.2007.051839. PMID 18039948.
- Sultan C, Biason-Lauber A, Philibert P (January 2009). "Mayer–Rokitansky–Kuster–Hauser syndrome: recent clinical and genetic findings". Gynecological Endocrinology. 25 (1): 8–11. doi:10.1080/09513590802288291. PMID 19165657.
- Wang X, Zhang X, Liu S, Li G, Cui L, Qin Y, Chen ZJ (December 2016). "Novel mutations in the TP63 gene are potentially associated with Müllerian duct anomalies". Human Reproduction. 31 (12): 2865–2871. doi:10.1093/humrep/dew259. PMID 27798044.
- Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ (August 2004). "A WNT4 mutation associated with Müllerian-duct regression and virilization in a 46,XX woman". The New England Journal of Medicine. 351 (8): 792–8. doi:10.1056/NEJMoa040533. PMID 15317892.
- Rackow BW, Arici A (June 2007). "Reproductive performance of women with müllerian anomalies". Current Opinion in Obstetrics & Gynecology. 19 (3): 229–37. doi:10.1097/GCO.0b013e32814b0649. PMID 17495638.
- Goldberg JM, Falcone T (July 1999). "Effect of diethylstilbestrol on reproductive function". Fertility and Sterility. 72 (1): 1–7. doi:10.1016/S0015-0282(99)00153-3. PMID 10428139.
- Mittendorf R (June 1995). "Teratogen update: carcinogenesis and teratogenesis associated with exposure to diethylstilbestrol (DES) in utero". Teratology. 51 (6): 435–45. doi:10.1002/tera.1420510609. PMID 7502243.
- Raga F, Bauset C, Remohi J, Bonilla-Musoles F, Simón C, Pellicer A (October 1997). "Reproductive impact of congenital Müllerian anomalies". Human Reproduction. 12 (10): 2277–81. doi:10.1093/humrep/12.10.2277. PMID 9402295.
- Cahen-Peretz A, Sheiner E, Friger M, Walfisch A (January 2019). "The association between Müllerian anomalies and perinatal outcome". The Journal of Maternal-Fetal & Neonatal Medicine. 32 (1): 51–57. doi:10.1080/14767058.2017.1370703. PMID 28826263.
- Reichman DE, Laufer MR (April 2010). "Congenital uterine anomalies affecting reproduction". Best Practice & Research. Clinical Obstetrics & Gynaecology. 24 (2): 193–208. doi:10.1016/j.bpobgyn.2009.09.006. PMID 19897423.
- Breech LL, Laufer MR (March 2009). "Müllerian anomalies". Obstetrics and Gynecology Clinics of North America. 36 (1): 47–68. doi:10.1016/j.ogc.2009.02.002. PMID 19344847.
- Fox NS, Roman AS, Stern EM, Gerber RS, Saltzman DH, Rebarber A (June 2014). "Type of congenital uterine anomaly and adverse pregnancy outcomes". The Journal of Maternal-Fetal & Neonatal Medicine. 27 (9): 949–53. doi:10.3109/14767058.2013.847082. PMID 24050215.
- Heinonen PK, Kuismanen K, Ashorn R (April 2000). "Assisted reproduction in women with uterine anomalies". European Journal of Obstetrics, Gynecology, and Reproductive Biology. 89 (2): 181–4. doi:10.1016/S0301-2115(99)00198-0. PMID 10725580.
- Attia KI, Hug-Koronya M, Ginsburg ES, Hornstein MD (October 2001). "Effects of Mullerian anomalies on in vitro fertilization outcome". Journal of Assisted Reproduction and Genetics. 18 (10): 544–7. doi:10.1023/A:1011950202480. PMC 3455312. PMID 11699126.
- Chan YY, Jayaprakasan K, Tan A, Thornton JG, Coomarasamy A, Raine-Fenning NJ (October 2011). "Reproductive outcomes in women with congenital uterine anomalies: a systematic review". Ultrasound in Obstetrics & Gynecology. 38 (4): 371–82. doi:10.1002/uog.10056. PMID 21830244.
- Marcus S, al-Shawaf T, Brinsden P (July 1996). "The obstetric outcome of in vitro fertilization and embryo transfer in women with congenital uterine malformation". American Journal of Obstetrics and Gynecology. 175 (1): 85–9. doi:10.1016/S0002-9378(96)70255-7. PMID 8694080.