Histone methylation
Histone methylation is a process by which methyl groups are transferred to amino acids of histone proteins that make up nucleosomes, which the DNA double helix wraps around to form chromosomes. Methylation of histones can either increase or decrease transcription of genes, depending on which amino acids in the histones are methylated, and how many methyl groups are attached. Methylation events that weaken chemical attractions between histone tails and DNA increase transcription because they enable the DNA to uncoil from nucleosomes so that transcription factor proteins and RNA polymerase can access the DNA. This process is critical for the regulation of gene expression that allows different cells to express different genes.
Function
Histone methylation, as a mechanism for modifying chromatin structure is associated with stimulation of neural pathways known to be important for formation of long-term memories and learning.[1] Animal models have shown methylation and other epigenetic regulation mechanisms to be associated with conditions of aging, neurodegenerative diseases, and intellectual disability[1] (Rubinstein–Taybi syndrome, X-linked intellectual disability).[2] This modification alters the properties of the nucleosome and affects its interactions with other proteins, particularly in regards to gene transcription processes.
- Histone methylation can be associated with either transcriptional repression or activation. For example, trimethylation of histone H3 at lysine 4 (H3K4me3) is an active mark for transcription and is upregulated in hippocampus one hour after contextual fear conditioning in rats. However, dimethylation of histone H3 at lysine 9 (H3K9me2), a signal for transcriptional silencing, is increased after exposure to either the fear conditioning or a novel environment alone.[3]
- Methylation of some lysine (K) and arginine (R) residues of histones results in transcriptional activation. Examples include methylation of lysine 4 of histone 3 (H3K4me1), and arginine (R) residues on H3 and H4.
- Addition of methyl groups to histones by histone methyltransferases, can either activate or further repress transcription, depending on the amino acid being methylated and the presence of other methyl or acetyl groups in the vicinity.[4]
Mechanism
The fundamental unit of chromatin, called a nucleosome, contains DNA wound around a protein octamer. This octamer consists of two copies each of four histone proteins: H2A, H2B, H3, and H4. Each one of these proteins has a tail extension, and these tails are the targets of nucleosome modification by methylation. DNA activation or inactivation is largely dependent on the specific tail residue methylated and its degree of methylation. Histones can be methylated on lysine (K) and arginine (R) residues only, but methylation is most commonly observed on lysine residues of histone tails H3 and H4.[5] The tail end furthest from the nucleosome core is the N-terminal (residues are numbered starting at this end). Common sites of methylation associated with gene activation include H3K4, H3K48, and H3K79. Common sites for gene inactivation include H3K9 and H3K27.[6] Studies of these sites have found that methylation of histone tails at different residues serve as markers for the recruitment of various proteins or protein complexes that serve to regulate chromatin activation or inactivation.
Lysine and arginine residues both contain amino groups, which confer basic and hydrophobic characteristics. Lysine is able to be mono-, di-, or trimethylated with a methyl group replacing each hydrogen of its NH3+ group. With a free NH2 and NH2+ group, arginine is able to be mono- or dimethylated. This dimethylation can occur asymmetrically on the NH2 group or symmetrically with one methylation on each group.[7] Each addition of a methyl group on each residue requires a specific set of protein enzymes with various substrates and cofactors. Generally, methylation of an arginine residue requires a complex including protein arginine methyltransferase (PRMT) while lysine requires a specific histone methyltransferase (HMT), usually containing an evolutionarily conserved SET domain.[8]
Different degrees of residue methylation can confer different functions, as exemplified in the methylation of the commonly studied H4K20 residue. Monomethylated H4K20 (H4K20me1) is involved in the compaction of chromatin and therefore transcriptional repression. However, H4K20me2 is vital in the repair of damaged DNA. When dimethylated, the residue provides a platform for the binding of protein 53BP1 involved in the repair of double-stranded DNA breaks by non-homologous end joining. H4K20me3 is observed to be concentrated in heterochromatin and reductions in this trimethylation are observed in cancer progression. Therefore, H4K20me3 serves an additional role in chromatin repression.[8] Repair of DNA double-stranded breaks in chromatin also occurs by homologous recombination and also involves histone methylation (H3K9me3) to facilitate access of the repair enzymes to the sites of damage.[9]
Histone methyltransferase
The genome is tightly condensed into chromatin, which needs to be loosened for transcription to occur. In order to halt the transcription of a gene the DNA must be wound tighter. This can be done by modifying histones at certain sites by methylation. Histone methyltransferases are enzymes which transfer methyl groups from S-Adenosyl methionine (SAM) onto the lysine or arginine residues of the H3 and H4 histones. There are instances of the core globular domains of histones being methylated as well.
The histone methyltransferases are specific to either lysine or arginine. The lysine-specific transferases are further broken down into whether or not they have a SET domain or a non-SET domain. These domains specify exactly how the enzyme catalyzes the transfer of the methyl from SAM to the transfer protein and further to the histone residue.[10] The methyltransferases can add 1-3 methyls on the target residues.
These methyls that are added to the histones act to regulate transcription by blocking or encouraging DNA access to transcription factors. In this way the integrity of the genome and epigenetic inheritance of genes are under the control of the actions of histone methyltransferases. Histone methylation is key in distinguishing the integrity of the genome and the genes that are expressed by cells, thus giving the cells their identities.
Methylated histones can either repress or activate transcription.[10] For example, while H3K4me2, H3K4me3, and H3K79me3 are generally associated with transcriptional activity, whereas H3K9me2, H3K9me3, H3K27me2, H3K27me3, and H4K20me3 are associated with transcriptional repression.[11]
Epigenetics
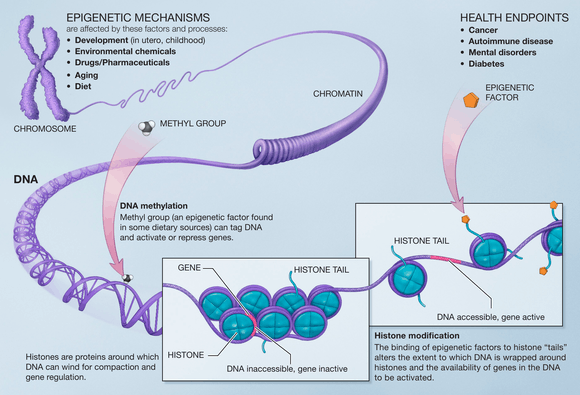
Modifications made on the histone have an effect on the genes that are expressed in a cell and this is the case when methyls are added to the histone residues by the histone methyltransferases.[12] Histone methylation plays an important role on the assembly of the heterochromatin mechanism and the maintenance of gene boundaries between genes that are transcribed and those that aren’t. These changes are passed down to progeny and can be affected by the environment that the cells are subject to. Epigenetic alterations are reversible meaning that they can be targets for therapy.
The activities of histone methyltransferases are offset by the activity of histone demethylases. This allows for the switching on or off of transcription by reversing pre-existing modifications. It is necessary for the activities of both histone methyltrasnsferases and histone demethylases to be regulated tightly. Misregulation of either can lead to gene expression that leads to increased susceptibility to disease. Many cancers arise from the inappropriate epigenetic effects of misregulated methylation.[13] However, because these processes are at times reversible, there is interest in utilizing their activities in concert with anti-cancer therapies.[13]
In X chromosome inactivation
In female organisms, a sperm containing an X chromosome fertilizes the egg, giving the embryo two copies of the X chromosome. Females, however, do not initially require both copies of the X chromosome as it would only double the amount of protein products transcribed as shown by the hypothesis of dosage compensation. The paternal X chromosome is quickly inactivated during the first few divisions.[14] This inactive X chromosome (Xi) is packed into an incredibly tight form of chromatin called heterochromatin.[15] This packing occurs due to the methylation of the different lysine residues that help form different histones. In humans X inactivation is a random process, that is mediated by the non-coding RNA XIST.[16]
Although methylation of lysine residues occurs on many different histones, the most characteristic of Xi occurs on the ninth lysine of the third histone (H3K9). While a single methylation of this region allows for the genes bound to remain transcriptionally active,[17] in heterochromatin this lysine residue is often methylated twice or three times, H3K9me2 or H3K9me3 respectively, to ensure that the DNA bound is inactive. More recent research has shown that H3K27me3 and H4K20me1 are also common in early embryos. Other methylation markings associated with transcriptionally active areas of DNA, H3K4me2 and H3K4me3, are missing from the Xi chromosome along with many acetylation markings. Although it was known that certain Xi histone methylation markings stayed relatively constant between species, it has recently been discovered that different organisms and even different cells within a single organism can have different markings for their X inactivation.[18] Through histone methylation, there is genetic imprinting, so that the same X homolog stays inactivated through chromosome replications and cell divisions.
Mutations
Due to the fact that histone methylation regulates much of what genes become transcribed, even slight changes to the methylation patterns can have dire effects on the organism. Mutations that occur to increase and decrease methylation have great changes on gene regulation, while mutations to enzymes such as methyltransferase and demethyltransferase can completely alter which proteins are transcribed in a given cell. Over methylation of a chromosome can cause certain genes that are necessary for normal cell function, to become inactivated. In a certain yeast strain, Saccharomyces cerevisiae, a mutation that causes three lysine residues on the third histone, H3K4, H3K36, and H3K79, to become methylated causes a delay in the mitotic cell cycle, as many genes required for this progression are inactivated. This extreme mutation leads to the death of the organism. It has been discovered that the deletion of genes that will eventually allow for the production of histone methyltransferase allows this organism to live as its lysine residues are not methylated.[19]
In recent years it has come to the attention of researchers that many types of cancer are caused largely due to epigenetic factors. Cancer can be caused in a variety of ways due to differential methylation of histones. Since the discovery of oncogenes as well as tumor suppressor genes it has been known that a large factor of causing and repressing cancer is within our own genome. If areas around oncogenes become unmethylated these cancer-causing genes have the potential to be transcribed at an alarming rate. Opposite of this is the methylation of tumor suppressor genes. In cases where the areas around these genes were highly methylated, the tumor suppressor gene was not active and therefore cancer was more likely to occur. These changes in methylation pattern are often due to mutations in methyltransferase and demethyltransferase.[20] Other types of mutations in proteins such as isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) can cause the inactivation of histone demethyltransferase which in turn can lead to a variety of cancers, gliomas and leukemias, depending on in which cells the mutation occurs.[21]
One-carbon metabolism modifies histone methylation
In one-carbon metabolism, the amino acids glycine and serine are converted via the folate and methionine cycles to nucleotide precursors and SAM. Multiple nutrients fuel one-carbon metabolism, including glucose, serine, glycine, and threonine. High levels of the methyl donor SAM influence histone methylation, which may explain how high SAM levels prevent malignant transformation.[22]
See also
References
- Kramer, Jamie M. (February 2013). "Epigenetic regulation of memory: implications in human cognitive disorders". Biomolecular Concepts. 4 (1): 1–12. doi:10.1515/bmc-2012-0026. hdl:2066/118379. PMID 25436561.
- Lilja, Tobias; Nina Heldring; Ola Hermanson (February 2013). "Like a rolling histone: Epigenetic regulation of neural stem cells and brain development by factors controlling histone acetylation and methylation". Biochimica et Biophysica Acta (BBA) - General Subjects. 1830 (2): 2354–2360. doi:10.1016/j.bbagen.2012.08.011. PMID 22986149.
- Gupta, Swati; Se Y. Kim; Sonja Artis; David L. Molfese; Armin Schumacher; J. David Sweatt; Richard E. Paylor; Farah D. Lubin (10 March 2010). "Histone Methylation Regulates Memory Formation". The Journal of Neuroscience. 30 (10): 3589–3599. doi:10.1523/JNEUROSCI.3732-09.2010. PMC 2859898. PMID 20219993.
- Greer, Eric L.; Shi, Yang (2012). "Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance". Nature Reviews Genetics. 13 (5): 343–57. doi:10.1038/nrg3173. PMC 4073795. PMID 22473383.
- Jia, Songtao; Wang, Yu (2009). "Degrees make all the difference" (PDF). Epigenetics. 4 (5): 1–4.
- Gilbert, S. F. (2010). Developmental biology. (9th ed.). Sinauer Associates, Inc. 35-37.
- Blanc, Roméo S; Richard, Stéphane (5 January 2017). "Arginine Methylation: The Coming of Age". Molecular Cell. 65 (1): 8–24. doi:10.1016/j.molcel.2016.11.003. PMID 28061334.
- Reinberg, Danny; Zhang, Yi (2001). "Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails". Genes & Development. 15 (18): 2343–2360. doi:10.1101/gad.927301. PMID 11562345.
- Wei S, Li C, Yin Z, Wen J, Meng H, Xue L, Wang J (2018). "Histone methylation in DNA repair and clinical practice: new findings during the past 5-years". J Cancer. 9 (12): 2072–2081. doi:10.7150/jca.23427. PMC 6010677. PMID 29937925.
- Rice, Judd C.; Briggs, Scott D.; Ueberheide, Beatrix; Barber, Cynthia M.; Shabanowitz, Jeffrey; Hunt, Donald F.; Shinkai, Yoichi; Allis, C.David (2003). "Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains". Molecular Cell. 12 (6): 1591–1598. doi:10.1016/S1097-2765(03)00479-9. PMID 14690610.
- Kooistra, Susanne Marije; Helin, Kristian (2012-04-04). "Molecular mechanisms and potential functions of histone demethylases". Nature Reviews Molecular Cell Biology. 13 (5): 297–311. doi:10.1038/nrm3327. ISSN 1471-0072. PMID 22473470.
- Cheung, Peter; Lau, Priscilla (2005). "Epigenetic Regulation by Histone Methylation and Histone Variants". Molecular Endocrinology. 19 (3): 563. doi:10.1210/me.2004-0496. PMID 15677708.
- Albert, Mareike; Helin, Kristian (2010). "Histone methyltransferases in cancer". Seminars in Cell & Developmental Biology. 21 (2): 209–220. doi:10.1016/j.semcdb.2009.10.007. PMID 19892027.
- Akagi, Nobuo; Sasaki, Motomichi (1975). "Preferential Inactivation of the Paternally Derived X Chromosome in the Extraembryonic Membranes of the Mouse". Nature. 256 (5519): 640–42. doi:10.1038/256640a0. PMID 1152998.
- Elgin, Sarah C.R.; Grewal, Shiv I.S. (2003). "Heterochromatin: Silence Is Golden". Current Biology. 13 (23): R895–898. doi:10.1016/j.cub.2003.11.006. PMID 14654010.
- Ahn, J.; Lee, J. (2008). "X chromosome: X inactivation". Nature Education. 1 (1): 24.
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.; Schones, D.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. (2007). "High-Resolution Profiling of Histone Methylations in the Human Genome". Cell. 129 (4): 823–37. doi:10.1016/j.cell.2007.05.009. PMID 17512414.
- Chow, Jennifer; Heard, Edith (2009). "X Inactivation and the Complexities of Silencing a Sex Chromosome". Current Opinion in Cell Biology. 21 (3): 359–66. doi:10.1016/j.ceb.2009.04.012. PMID 19477626.
- Jin, Y.; Rodriguez, A. M.; Stanton, J. D.; Kitazono, A. A.; Wyrick, J. J. (2007). "Simultaneous Mutation of Methylated Lysine Residues in Histone H3 Causes Enhanced Gene Silencing, Cell Cycle Defects, and Cell Lethality in Saccharomyces Cerevisiae". Molecular and Cellular Biology. 27 (19): 6832–841. doi:10.1128/mcb.00745-07. PMC 2099221. PMID 17664279.
- Esteller, M (2006). "Epigenetics Provides a New Generation of Oncogenes and Tumour-suppressor Genes". British Journal of Cancer. 94 (2): 179–83. doi:10.1038/sj.bjc.6602918. PMC 2361113. PMID 16404435.
- Lu, Chao; Ward, Patrick S.; Kapoor, Gurpreet S.; Rohle, Dan; Turcan, Sevin; Abdel-Wahab, Omar; Edwards, Christopher R.; Khanin, Raya; Figueroa, Maria E.; Melnick, Ari; Wellen, Kathryn E.; O'Rourke, Donald M.; Berger, Shelley L.; Chan, Timothy A.; Levine, Ross L.; Mellinghoff, Ingo K.; Thompson, Craig B. (2012). "DH Mutation Impairs Histone Demethylation and Results in a Block to Cell Differentiation". Nature. 483 (7390): 474–78. doi:10.1038/nature10860. PMC 3478770. PMID 22343901.
- Wang YP, Lei QY (2018). "Metabolic recoding of epigenetics in cancer". Cancer Commun (Lond). 38 (1): 25. doi:10.1186/s40880-018-0302-3. PMC 5993135. PMID 29784032.
Further reading
- Orouji, Elias & Utikal, Jochen. (2018). Tackling malignant melanoma epigenetically: histone lysine methylation. Clinical Epigenetics 2018 10:145 https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-018-0583-z
- Gozani, O., & Shi, Y. (2014). Histone Methylation in Chromatin Signaling. In: Fundamentals of Chromatin (pp. 213–256). Springer New York. doi: 10.1007/978-1-4614-8624-4_5
- Shi, Yang; Whetstine, Johnathan R. (January 12, 2007). "Dynamic Regulation of Histone Lysine Methylation by Demethylases". Molecular Cell. 25 (1): 1–14. doi:10.1016/j.molcel.2006.12.010. PMID 17218267.