Genome skimming
Genome skimming is a sequencing approach that uses low-pass, shallow sequencing of a genome (up to 5%), to generate fragments of DNA, known as genome skims.[1][2] These genome skims contain information about the high-copy fraction of the genome.[2] The high-copy fraction of the genome consists of the ribosomal DNA, plastid genome (plastome), mitochondrial genome (mitogenome), and nuclear repeats such as microsatellites and transposable elements.[3] It employs high-throughput, next generation sequencing technology to generate these skims.[1] Although these skims are merely 'the tip of the genomic iceberg', phylogenomic analysis of them can still provide insights on evolutionary history and biodiversity at a lower cost and larger scale than traditional methods.[2][3][4] Due to the small amount of DNA required for genome skimming, its methodology can be applied in other fields other than genomics. Tasks like this include determining the traceability of products in the food industry, enforcing international regulations regarding biodiversity and biological resources, and forensics.[5]
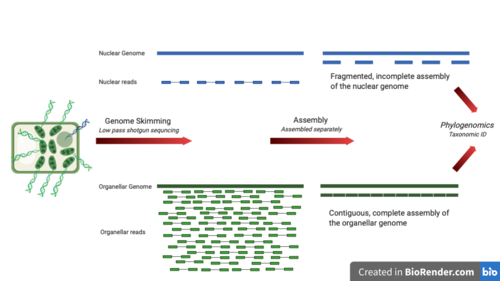
Current Uses
In addition to the assembly of the smaller organellar genomes, genome skimming can also be used to uncover conserved ortholog sequences for phylogenomic studies. In phylogenomic studies of multicellular pathogens, genome skimming can be used to find effector genes, discover endosymbionts and characterize genomic variation.[6]
High-copy DNA
Ribosomal DNA
The Internal transcribed spacers (ITS) are non-coding regions within the 18-5.8-28S rDNA in eukaryotes, and are one feature of rDNA that has been used in genome skimming studies.[7] ITS are used to detect different species within a genus, due to their high inter-species variability.[7] These have low individual variability, preventing identification of distinct strains or individuals.[7] They are also present in all eukaryotes, has a high evolution rate, and has been used in phylogenetic analysis between and across species.[7]
When targeting nuclear rDNA, it is suggested that a minimum final sequencing depth of 100X is achieved, and sequences with less than 5X depth are masked.[1]
Plastomes
The plastid genome, or plastome, has been used extensively in identification and evolutionary studies using genome skimming due to its high abundance within plants (~3-5% of cell DNA), small size, simple structure, greater conservation of gene structure than nuclear or mitochondrial genes.[8][9] Plastids studies have previously been limited by the number of regions that could be assessed in traditional approaches.[9] Using genome skimming, the sequencing of the entire plastid genome, or plastome, can be done at a fraction of the cost and time required for typical sequencing approaches like Sanger sequencing.[3] Plastomes have been suggested as a method to replace traditional DNA barcodes in plants,[3] such as the rbcL and matK barcode genes. Compared to the typical DNA barcode, genome skimming produces plastomes at a tenth of the cost per base.[5] Recent uses of genome skims of plastomes have allowed greater resolution of phylogenies, higher differentiation of specific groups within taxa, and more accurate estimates of biodiversity.[9] Additionally, the plastome has been used to compare species within a genus to look at evolutionary changes and diversity within a group.[9]
When targeting plastomes, it is suggested that a minimum final sequencing depth of 30X is achieved for single-copy regions to ensure high quality assemblies. Single nucleotide polymorphisms (SNPs) with less than 20X depth should be masked.[1]
Mitogenomes
The mitochondrial genome, or mitogenome, is used as a molecular marker in a great variety of studies because of its maternal inheritance, high copy-number in the cell, lack of recombination, and high mutation rate. It’s often used for phylogenetic studies as it is very uniform across metazoan groups, with a circular, double-stranded DNA molecule structure, about 15 to 20 kilobases, with 37 ribosomal RNA genes, 13 protein-coding genes, and 22 transfer RNA genes. Mitochondrial barcode sequences, such as COI, NADH2, 16S rRNA, and 12S rRNA, can also be used for taxonomic identification.[10] The increased publishing of complete mitogenomes allows for inference of robust phylogenies across many taxonomic groups, and it can capture events such as gene rearrangements and positioning of mobile genetic elements. Using genome skimming to assemble complete mitogenomes, phylogenetic history and biodiversity of many organisms can be resolved.[4]
When targeting mitogenomes, there are no specific suggestions for minimum final sequencing depth, as mitogenomes are more variable in size and more variable in complexity in plant species, increasing the difficulty of assembling repeated sequences. However, highly conserved coding sequences and nonrepetitive flanking regions can be assembled using reference-guided assembly. Sequences should be masked similarly to targeting plastomes and nuclear ribosomal DNA.[1]
Nuclear repeats (satellites or transposable elements)
Nuclear repeats in the genome are an underused source of phylogenetic data. When the nuclear genome is sequenced at 5% of the genome, thousands of copies of the nuclear repeats will be present. Although the repeats sequenced will only be representative of those in the entire genome, it has been shown that these sequenced fractions accurately reflect genomic abundance. These repeats can be clustered de novo and their abundance is estimated. The distribution and occurrence of these repeat types can be phylogenetically informative and provide information about the evolutionary history of various species.[1]
Low-copy DNA
Low-copy DNA can prove useful for evolution developmental and phylogenetic studies.[11] It can be mined from high-copy fractions in a number of ways such as developing primers from databases that contain conserved orthologous genes, single‐copy conserved orthologous gene, and shared copy genes.[11] Another method is looking for novel probes that target low copy genes using transcriptomics via Hyb-Seq.[11] While nuclear genomes assembled using genome skims are extremely fragmented, some low-copy single-copy nuclear genes can be successfully assembled.[12]
Low-quantity degraded DNA
Previous methods of trying to recover degraded DNA were based on Sanger sequencing and relied on large intact DNA templates and were affected by contamination and method of preservation. Genome skimming, on the other hand, can be used to extract genetic information from preserved species in herbariums and museums, where the DNA is often very degraded, and very little remains.[4][13] Studies in plants show that DNA as old as 80 years and with as little as 500 pg of degraded DNA, can be used with genome skimming to infer genomic information.[13] In herbaria, even with low yield and low quality DNA, one study was still able to produce "high-quality complete chloroplast and ribosomal DNA sequences" at a large scale for downstream analyses.[14]
In field studies, invertebrates are stored in ethanol which is usually discarded during DNA-based studies.[15] Genome skimming has been shown to detect the low quantity of DNA from this ethanol-fraction and provide information about the biomass of the specimens in a fraction, the microbiota of outer tissue layers and the gut contents (like prey) released by the vomit reflex.[15] Thus, genome skimming can provide an additional method of understanding ecology via low copy DNA.[15]
Workflow
DNA extraction
DNA extraction protocols will vary depending on the source of the sample (i.e. plants, animals, etc.). The following DNA extraction protocols have been used in genome skimming:
Plants
|
Other
|
Library preparation
Library preparation protocols will depend on a variety of factors: organism, tissue type, etc. In the cases of preserved specimens, specific library preparation protocols modifications may have to be made.[1] The following library preparation protocols have been used in genome skimming:
Sequencing
Sequencing with short reads or long reads will depend on the target genome or genes. Microsatellites in nuclear repeats require longer reads.[23] The following sequencing platforms have been used in genome skimming:
- Illumina HiSeq 2000 platform[5][18][24][25]
- Illumina HiSeq 2500 platform[8][9][14][20][17][26]
- Illumina HiSeq 4000 platform[19]
- Illumina HiSeq X Ten platform[7][13][19]
- Illumina MiSeq platform[4][6][15][16][21][23]
- Illumina NextSeq 550 platform[4][21]
- Illumina GAIIx platform[1]
- Oxford Nanopore Technologies (ONT) MinION[10]
The Illumina MiSeq platform has been chosen by certain researchers for its long read length for short reads.[6]
Assembly
After genome skimming, high-copy organellar DNA can be assembled with a reference guide or assembled de novo. High-copy nuclear repeats can be clustered de novo.[1] Assemblers chosen will depend on the target genome and whether short or long reads are used. The following tools have been used to assemble genomes from genome skims:
Other
Annotation
Annotation is used to identify genes in the genome assemblies. The annotation tool chosen will depend on the target genome, and the target features of that genome. The following annotation tools have been used in genome skimming to annotate organellar genomes:
Other
Phylogeny construction
The assembled sequences are globally aligned, and then phylogenetic trees are constructed using phylogeny construction software. The software chosen for phylogeny construction will depend on whether a Maximum Likelihood (ML), Maximum Parsimony (MP), or Bayesian Inference (BI) method is appropriate. The following phylogeny construction programs have been used in genome skimming:
Tools and Pipelines
Various protocols, pipelines, and bioinformatic tools have been developed to help automate the downstream processes of genome skimming.
Hyb-Seq
Hyb-Seq is a new protocol for capturing low-copy nuclear genes that combines target enrichment and genome skimming.[29] Target enrichment of the low-copy loci is achieved through designed enrichment probes for specific single-copy exons, but requires a nuclear draft genome and transcriptome of the targeted organism. The target-enriched libraries are then sequenced, and the resulting reads processed, assembled, and identified. Using off-target reads, rDNA cistrons and complete plastomes can also be assembled. Through this process, Hyb-Seq is able to produce genome-scale datasets for phylogenomics.
GetOrganelle
GetOrganelle is a toolkit that assembles organellar genomes uses genome skimming reads.[30] Organelle-associated reads are recruited using a modified “baiting and iterative mapping” approach. The reads aligning to the target genome, using Bowtie2,[31] are referred to as “seed reads”. The seed reads are used as “baits” to recruit more organelle-associated reads via multiple iterations of extension. The read extension algorithm uses a hashing approach, where the reads are cut into substrings of certain lengths, referred to as “words”. At each extension iteration, these “words” are added to a hash table, referred to as a “baits pool”, which dynamically increases in size with each iteration. Due to the low sequencing coverage of genome skims, non-target reads, even those with high sequence similarity to target reads, are largely not recruited. Using the final recruited organellar-associated reads, GetOrganelle conducts a de novo assembly, using SPAdes.[32] The assembly graph is filtered and untangled, producing all possible paths of the graph, and therefore all configurations of the circular organellar genomes.
Skmer
Skmer is an assembly-free and alignment-free tool to compute genomic distances between the query and reference genome skims.[33] Skmer uses a 2 stage approach to compute these distances. First, it generates k-mer frequency profiling using a tool called JellyFish[34] and then these k-mers are converted into hashes.[33] A random subset of these hashes are selected to form a so-called "sketch".[33] For its second stage, Skmer uses Mash[35] to estimate the Jaccard index of two of these sketches.[33] The combination of these 2 stages is used to estimate the evolutionary distance.[33]
In silico Genome skimming
Although genome skimming is usually chosen as a cost-effective method to sequence organellar genomes, genome skimming can be done in silico if (deep) whole-genome sequencing data has already been obtained. Genome skimming has been demonstrated to simplify organellar genome assembly by subsampling the reads of the nuclear genome via in silico genome skimming.[36][37] Since the organellar genomes will be high-copy in the cell, in silico genome skimming essentially filters out nuclear sequences, leaving a higher organellar to nuclear sequence ratio for assembly, reducing the complexity of the assembly paradigm. In silico genome skimming was first done as a proof-of-concept, optimizing the parameters for read type, read length, and sequencing coverage.[1]
Other Applications
Other than the current uses listed above, genome skimming has also been applied to other tasks, such as quantifying pollen mixtures,[19] monitoring and conservation of certain populations.[38] Genome skimming can also be used for variant calling, to examine single nucleotide polymorphisms across a species.[22]
Advantages
Genome skimming is a cost-effective, rapid and reliable method to generate large shallow datasets,[5] since several datasets (plastid, mitochondrial, nuclear) are generated per run.[3] It is very simple to implement, requires less lab work and optimization, and does not require a priori knowledge of the organism nor its genome size.[3] This provides a low-risk avenue for biological inquiry and hypothesis generation without a huge commitment of resources.[6]
Genome skimming is an especially advantageous approach regarding cases where the genomic DNA may be old and degraded from chemical treatments, such as specimens from herbarium and museum collections,[4] a largely untapped genomic resource. Genome skimming allows for the molecular characterization of rare or extinct species.[5] The preservation processes in ethanol often damage the genomic DNA, which hinders the success of standard PCR protocols[3] and other amplicon-based approaches.[5] This presents an opportunity to sequence samples with very low DNA concentrations, without the need for DNA enrichment or amplification. Library preparation for specific to genome skimming has been shown to work with as low as 37 ng of DNA (0.2 ng/ul), 135-fold less than recommended by Illumina.[1]
Although genome skimming is mostly used to extract high-copy plastomes and mitogenomes, it can also provide partial sequences of low-copy nuclear sequences. These sequences may not be sufficiently complete for phylogenomic analysis, but can be sufficient for designing PCR primers and probes for hybridization-based approaches.[1]
Genome skimming is not dependent on any specific primers and remains unaffected by gene rearrangements.[4]
Limitations
Genome skimming scratches the surface of the genome, so it will not suffice for biological questions that require gene prediction and annotation.[6] These downstream steps are required for deep and more meaningful analyses.
Although plastid genomic sequences are abundant in genome skims, the presence of mitochondrial and nuclear pseudogenes of plastid origin can potentially pose issues for plastome assemblies.[1]
A combination of sequencing depth and read type, as well as genomic target (plastome, mitogenome, etc.) will influence the success of single-end and paired-end assemblies, so these parameters must be carefully chosen.[1]
Scalability
Both the wet-lab and the bioinformatics parts of genome skimming have certain challenges with scalability. Although the cost of sequencing in genome skimming is affordable at $80 for 1 Gb in 2016, the library preparation for sequencing is still very expensive, at least ~$200 per sample (as of 2016). Additionally, most library preparation protocols have not been fully automated with robotics yet. On the bioinformatics side, large complex databases and automated workflows need to be designed to handle the large amounts of data resulting from genome skimming. The automation of the following processes need to be implemented:[39]
- Assembly of the standard barcodes
- Assembly of organellar DNA (as well as nuclear ribosomal tandem repeats)
- Annotation of the different assembled fragments
- Removal of potential contaminant sequences
- Estimation of sequencing coverage for single-copy genes
- Extraction of reads corresponding to single-copy genes
- Identification of unknown specimen from a small shotgun sequencing or any DNA fragment
- Identification of the different organisms from shotgun sequencing of environmental DNA (metagenomics)
Some of these scalability challenges have already been implemented, as shown above in the "Tools and Pipelines" section.
See also
References
- Straub, Shannon C. K.; Parks, Matthew; Weitemier, Kevin; Fishbein, Mark; Cronn, Richard C.; Liston, Aaron (February 2012). "Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics". American Journal of Botany. 99 (2): 349–364. doi:10.3732/ajb.1100335. PMID 22174336.
- Dodsworth, Steven (September 2015). "Genome skimming for next-generation biodiversity analysis". Trends in Plant Science. 20 (9): 525–527. doi:10.1016/j.tplants.2015.06.012. PMID 26205170.
- Dodsworth, Steven Andrew, author. Genome skimming for phylogenomics. OCLC 1108700470.CS1 maint: multiple names: authors list (link)
- Trevisan, Bruna; Alcantara, Daniel M.C.; Machado, Denis Jacob; Marques, Fernando P.L.; Lahr, Daniel J.G. (2019-09-13). "Genome skimming is a low-cost and robust strategy to assemble complete mitochondrial genomes from ethanol preserved specimens in biodiversity studies". PeerJ. 7: e7543. doi:10.7717/peerj.7543. ISSN 2167-8359. PMC 6746217. PMID 31565556.
- Malé, Pierre-Jean G.; Bardon, Léa; Besnard, Guillaume; Coissac, Eric; Delsuc, Frédéric; Engel, Julien; Lhuillier, Emeline; Scotti-Saintagne, Caroline; Tinaut, Alexandra; Chave, Jérôme (April 2014). "Genome skimming by shotgun sequencing helps resolve the phylogeny of a pantropical tree family". Molecular Ecology Resources. 14 (5): 966–75. doi:10.1111/1755-0998.12246. PMID 24606032.
- Denver, Dee R.; Brown, Amanda M. V.; Howe, Dana K.; Peetz, Amy B.; Zasada, Inga A. (2016-08-04). Round, June L. (ed.). "Genome Skimming: A Rapid Approach to Gaining Diverse Biological Insights into Multicellular Pathogens". PLOS Pathogens. 12 (8): e1005713. doi:10.1371/journal.ppat.1005713. ISSN 1553-7374. PMC 4973915. PMID 27490201.
- Lin, Geng-Ming; Lai, Yu-Heng; Audira, Gilbert; Hsiao, Chung-Der (November 2017). "A Simple Method to Decode the Complete 18-5.8-28S rRNA Repeated Units of Green Algae by Genome Skimming". International Journal of Molecular Sciences. 18 (11): 2341. doi:10.3390/ijms18112341. PMC 5713310. PMID 29113146.
- Liu, Luxian; Wang, Yuewen; He, Peizi; Li, Pan; Lee, Joongku; Soltis, Douglas E.; Fu, Chengxin (2018-04-04). "Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data". BMC Genomics. 19 (1): 235. doi:10.1186/s12864-018-4633-x. ISSN 1471-2164. PMC 5885378. PMID 29618324.
- Hinsinger, Damien Daniel; Strijk, Joeri Sergej (2019-01-10). "Plastome of Quercus xanthoclada and comparison of genomic diversity amongst selected Quercus species using genome skimming". PhytoKeys. 132: 75–89. doi:10.3897/phytokeys.132.36365. ISSN 1314-2003. PMC 6783484. PMID 31607787.
- Johri, Shaili; Solanki, Jitesh; Cantu, Vito Adrian; Fellows, Sam R.; Edwards, Robert A.; Moreno, Isabel; Vyas, Asit; Dinsdale, Elizabeth A. (December 2019). "'Genome skimming' with the MinION hand-held sequencer identifies CITES-listed shark species in India's exports market". Scientific Reports. 9 (1): 4476. Bibcode:2019NatSR...9.4476J. doi:10.1038/s41598-019-40940-9. ISSN 2045-2322. PMC 6418218. PMID 30872700.
- Berger, Brent A.; Han, Jiahong; Sessa, Emily B.; Gardner, Andrew G.; Shepherd, Kelly A.; Ricigliano, Vincent A.; Jabaily, Rachel S.; Howarth, Dianella G. (2017). "The unexpected depths of genome-skimming data: A case study examining Goodeniaceae floral symmetry genes1". Applications in Plant Sciences. 5 (10): 1700042. doi:10.3732/apps.1700042. ISSN 2168-0450. PMC 5664964. PMID 29109919.
- Berger, Brent A.; Han, Jiahong; Sessa, Emily B.; Gardner, Andrew G.; Shepherd, Kelly A.; Ricigliano, Vincent A.; Jabaily, Rachel S.; Howarth, Dianella G. (October 2017). "The Unexpected Depths of Genome-Skimming Data: A Case Study Examining Goodeniaceae Floral Symmetry Genes". Applications in Plant Sciences. 5 (10): 1700042. doi:10.3732/apps.1700042. ISSN 2168-0450. PMC 5664964. PMID 29109919.
- Zeng, Chun-Xia; Hollingsworth, Peter M.; Yang, Jing; He, Zheng-Shan; Zhang, Zhi-Rong; Li, De-Zhu; Yang, Jun-Bo (2018-06-05). "Genome skimming herbarium specimens for DNA barcoding and phylogenomics". Plant Methods. 14 (1): 43. doi:10.1186/s13007-018-0300-0. ISSN 1746-4811. PMC 5987614. PMID 29928291.
- Nevill, Paul G.; Zhong, Xiao; Tonti-Filippini, Julian; Byrne, Margaret; Hislop, Michael; Thiele, Kevin; van Leeuwen, Stephen; Boykin, Laura M.; Small, Ian (2020-01-04). "Large scale genome skimming from herbarium material for accurate plant identification and phylogenomics". Plant Methods. 16 (1): 1. doi:10.1186/s13007-019-0534-5. ISSN 1746-4811. PMC 6942304. PMID 31911810.
- Linard, B.; Arribas, P.; Andújar, C.; Crampton‐Platt, A.; Vogler, A. P. (2016). "Lessons from genome skimming of arthropod-preserving ethanol" (PDF). Molecular Ecology Resources. 16 (6): 1365–1377. doi:10.1111/1755-0998.12539. hdl:10044/1/49937. ISSN 1755-0998. PMID 27235167.
- Liu, Shih-Hui; Edwards, Christine E.; Hoch, Peter C.; Raven, Peter H.; Barber, Janet C. (May 2018). "Genome skimming provides new insight into the relationships in Ludwigia section Macrocarpon, a polyploid complex". American Journal of Botany. 105 (5): 875–887. doi:10.1002/ajb2.1086. PMID 29791715.
- Nauheimer, Lars; Cui, Lujing; Clarke, Charles; Crayn, Darren M.; Bourke, Greg; Nargar, Katharina (2019). "Genome skimming provides well resolved plastid and nuclear phylogenies, showing patterns of deep reticulate evolution in the tropical carnivorous plant genus Nepenthes (Caryophyllales)". Australian Systematic Botany. 32 (3): 243–254. doi:10.1071/SB18057. ISSN 1030-1887.
- Ripma, Lee A.; Simpson, Michael G.; Hasenstab-Lehman, Kristen (December 2014). "Geneious! Simplified Genome Skimming Methods for Phylogenetic Systematic Studies: A Case Study in Oreocarya (Boraginaceae)". Applications in Plant Sciences. 2 (12): 1400062. doi:10.3732/apps.1400062. ISSN 2168-0450. PMC 4259456. PMID 25506521.
- Lang, Dandan; Tang, Min; Hu, Jiahui; Zhou, Xin (November 2019). "Genome‐skimming provides accurate quantification for pollen mixtures". Molecular Ecology Resources. 19 (6): 1433–1446. doi:10.1111/1755-0998.13061. ISSN 1755-098X. PMC 6900181. PMID 31325909.
- Stoughton, Thomas R.; Kriebel, Ricardo; Jolles, Diana D.; O'Quinn, Robin L. (March 2018). "Next-generation lineage discovery: A case study of tuberous Claytonia L." American Journal of Botany. 105 (3): 536–548. doi:10.1002/ajb2.1061. PMID 29672830.
- Dodsworth, Steven; Guignard, Maïté S.; Christenhusz, Maarten J. M.; Cowan, Robyn S.; Knapp, Sandra; Maurin, Olivier; Struebig, Monika; Leitch, Andrew R.; Chase, Mark W.; Forest, Félix (2018-10-29). "Potential of Herbariomics for Studying Repetitive DNA in Angiosperms". Frontiers in Ecology and Evolution. 6: 174. doi:10.3389/fevo.2018.00174. ISSN 2296-701X.
- Jackson, David; Emslie, Steven D; van Tuinen, Marcel (2012). "Genome skimming identifies polymorphism in tern populations and species". BMC Research Notes. 5 (1): 94. doi:10.1186/1756-0500-5-94. ISSN 1756-0500. PMC 3292991. PMID 22333071.
- Xia, Yun; Luo, Wei; Yuan, Siqi; Zheng, Yuchi; Zeng, Xiaomao (December 2018). "Microsatellite development from genome skimming and transcriptome sequencing: comparison of strategies and lessons from frog species". BMC Genomics. 19 (1): 886. doi:10.1186/s12864-018-5329-y. ISSN 1471-2164. PMC 6286531. PMID 30526480.
- Fonseca, Luiz Henrique M.; Lohmann, Lúcia G. (January 2020). "Exploring the potential of nuclear and mitochondrial sequencing data generated through genome‐skimming for plant phylogenetics: A case study from a clade of neotropical lianas". Journal of Systematics and Evolution. 58 (1): 18–32. doi:10.1111/jse.12533. ISSN 1674-4918.
- Bock, Dan G.; Kane, Nolan C.; Ebert, Daniel P.; Rieseberg, Loren H. (February 2014). "Genome skimming reveals the origin of the Jerusalem Artichoke tuber crop species: neither from Jerusalem nor an artichoke". New Phytologist. 201 (3): 1021–1030. doi:10.1111/nph.12560. PMID 24245977.
- Richter, Sandy; Schwarz, Francine; Hering, Lars; Böggemann, Markus; Bleidorn, Christoph (December 2015). "The Utility of Genome Skimming for Phylogenomic Analyses as Demonstrated for Glycerid Relationships (Annelida, Glyceridae)". Genome Biology and Evolution. 7 (12): 3443–3462. doi:10.1093/gbe/evv224. ISSN 1759-6653. PMC 4700955. PMID 26590213.
- Grandjean, Frederic; Tan, Mun Hua; Gan, Han Ming; Lee, Yin Peng; Kawai, Tadashi; Distefano, Robert J.; Blaha, Martin; Roles, Angela J.; Austin, Christopher M. (November 2017). "Rapid recovery of nuclear and mitochondrial genes by genome skimming from Northern Hemisphere freshwater crayfish". Zoologica Scripta. 46 (6): 718–728. doi:10.1111/zsc.12247.
- "Geneious – OSTR". Retrieved 2020-02-28.
- Weitemier, Kevin; Straub, Shannon C. K.; Cronn, Richard C.; Fishbein, Mark; Schmickl, Roswitha; McDonnell, Angela; Liston, Aaron (September 2014). "Hyb-Seq: Combining Target Enrichment and Genome Skimming for Plant Phylogenomics". Applications in Plant Sciences. 2 (9): 1400042. doi:10.3732/apps.1400042. ISSN 2168-0450. PMC 4162667. PMID 25225629.
- Jin, Jian-Jun; Yu, Wen-Bin; Yang, Jun-Bo; Song, Yu; dePamphilis, Claude W.; Yi, Ting-Shuang; Li, De-Zhu (2018-03-09). "GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes". doi:10.1101/256479. Cite journal requires
|journal=(help) - Langmead, Ben; Salzberg, Steven L (Mar 2012). "Fast gapped-read alignment with Bowtie 2". Nature Methods. 9 (4): 357–359. doi:10.1038/nmeth.1923. ISSN 1548-7091. PMC 3322381. PMID 22388286.
- Bankevich, Anton; Nurk, Sergey; Antipov, Dmitry; Gurevich, Alexey A.; Dvorkin, Mikhail; Kulikov, Alexander S.; Lesin, Valery M.; Nikolenko, Sergey I.; Pham, Son; Prjibelski, Andrey D.; Pyshkin, Alexey V. (May 2012). "SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing". Journal of Computational Biology. 19 (5): 455–477. doi:10.1089/cmb.2012.0021. ISSN 1066-5277. PMC 3342519. PMID 22506599.
- Sarmashghi, Shahab; Bohmann, Kristine; P. Gilbert, M. Thomas; Bafna, Vineet; Mirarab, Siavash (December 2019). "Skmer: assembly-free and alignment-free sample identification using genome skims". Genome Biology. 20 (1): 34. doi:10.1186/s13059-019-1632-4. ISSN 1474-760X. PMC 6374904. PMID 30760303.
- Marçais, Guillaume; Kingsford, Carl (2011-03-15). "A fast, lock-free approach for efficient parallel counting of occurrences of k-mers". Bioinformatics. 27 (6): 764–770. doi:10.1093/bioinformatics/btr011. ISSN 1460-2059. PMC 3051319. PMID 21217122.
- Ondov, Brian D.; Treangen, Todd J.; Melsted, Páll; Mallonee, Adam B.; Bergman, Nicholas H.; Koren, Sergey; Phillippy, Adam M. (Dec 2016). "Mash: fast genome and metagenome distance estimation using MinHash". Genome Biology. 17 (1): 132. doi:10.1186/s13059-016-0997-x. ISSN 1474-760X. PMC 4915045. PMID 27323842.
- Lin, Diana; Coombe, Lauren; Jackman, Shaun D.; Gagalova, Kristina K.; Warren, René L.; Hammond, S. Austin; Kirk, Heather; Pandoh, Pawan; Zhao, Yongjun; Moore, Richard A.; Mungall, Andrew J. (2019-06-06). Rokas, Antonis (ed.). "Complete Chloroplast Genome Sequence of a White Spruce ( Picea glauca, Genotype WS77111) from Eastern Canada". Microbiology Resource Announcements. 8 (23): e00381–19, /mra/8/23/MRA.00381–19.atom. doi:10.1128/MRA.00381-19. ISSN 2576-098X. PMC 6554609. PMID 31171622.
- Lin, Diana; Coombe, Lauren; Jackman, Shaun D.; Gagalova, Kristina K.; Warren, René L.; Hammond, S. Austin; McDonald, Helen; Kirk, Heather; Pandoh, Pawan; Zhao, Yongjun; Moore, Richard A. (2019-06-13). Stajich, Jason E. (ed.). "Complete Chloroplast Genome Sequence of an Engelmann Spruce ( Picea engelmannii, Genotype Se404-851) from Western Canada". Microbiology Resource Announcements. 8 (24): e00382–19, /mra/8/24/MRA.00382–19.atom. doi:10.1128/MRA.00382-19. ISSN 2576-098X. PMC 6588038. PMID 31196920.
- Johri, Shaili; Doane, Michael; Allen, Lauren; Dinsdale, Elizabeth (2019-03-29). "Taking Advantage of the Genomics Revolution for Monitoring and Conservation of Chondrichthyan Populations". Diversity. 11 (4): 49. doi:10.3390/d11040049. ISSN 1424-2818.
- Coissac, Eric; Hollingsworth, Peter M.; Lavergne, Sébastien; Taberlet, Pierre (April 2016). "From barcodes to genomes: extending the concept of DNA barcoding". Molecular Ecology. 25 (7): 1423–1428. doi:10.1111/mec.13549. PMID 26821259.
| Key components | |
|---|---|
| Fields | |
| Archaeogenetics of | |
| Related topics | |
| Lists | |
| |