Gel electrophoresis of nucleic acids
Nucleic acid electrophoresis is an analytical technique used to separate DNA or RNA fragments by size and reactivity. Nucleic acid molecules which are to be analyzed are set upon a viscous medium, the gel, where an electric field induces the nucleic acids (which are negatively charged due to their sugar-phosphate backbone) to migrate toward the anode (which is positively charged because this is an electrolytic rather than galvanic cell). The separation of these fragments is accomplished by exploiting the mobilities with which different sized molecules are able to pass through the gel. Longer molecules migrate more slowly because they experience more resistance within the gel. Because the size of the molecule affects its mobility, smaller fragments end up nearer to the anode than longer ones in a given period. After some time, the voltage is removed and the fragmentation gradient is analyzed. For larger separations between similar sized fragments, either the voltage or run time can be increased. Extended runs across a low voltage gel yield the most accurate resolution. Voltage is, however, not the sole factor in determining electrophoresis of nucleic acids.
The nucleic acid to be separated can be prepared in several ways before separation by electrophoresis. In the case of large DNA molecules, the DNA is frequently cut into smaller fragments using a DNA restriction endonuclease (or restriction enzyme). In other instances, such as PCR amplified samples, enzymes present in the sample that might affect the separation of the molecules are removed through various means before analysis. Once the nucleic acid is properly prepared, the samples of the nucleic acid solution are placed in the wells of the gel and a voltage is applied across the gel for a specified amount of time.
The DNA fragments of different lengths are visualized using a fluorescent dye specific for DNA, such as ethidium bromide. The gel shows bands corresponding to different nucleic acid molecules populations with different molecular weight. Fragment size is usually reported in "nucleotides", "base pairs" or "kb" (for thousands of base pairs) depending upon whether single- or double-stranded nucleic acid has been separated. Fragment size determination is typically done by comparison to commercially available DNA markers containing linear DNA fragments of known length.
The types of gel most commonly used for nucleic acid electrophoresis are agarose (for relatively long DNA molecules) and polyacrylamide (for high resolution of short DNA molecules, for example in DNA sequencing). Gels have conventionally been run in a "slab" format such as that shown in the figure, but capillary electrophoresis has become important for applications such as high-throughput DNA sequencing. Electrophoresis techniques used in the assessment of DNA damage include alkaline gel electrophoresis and pulsed field gel electrophoresis.
For short DNA segments such as 20 to 60 bp double stranded DNA, running them in polyacrylamide gel (PAGE) will give better resolution (native condition).[1] Similarly, RNA and single-stranded DNA can be run and visualised by PAGE gels containing denaturing agents such as Urea. PAGE gels are widely used in techniques such as DNA foot printing, EMSA and other DNA-protein interaction techniques.
The measurement and analysis are mostly done with a specialized gel analysis software. Capillary electrophoresis results are typically displayed in a trace view called an electropherogram.
Factors affecting migration of nucleic acids
A number of factors can affect the migration of nucleic acids: the dimension of the gel pores, the voltage used, the ionic strength of the buffer, and the concentration intercalating dye such as ethidium bromide if used during electrophoresis.[2]
Size of DNA
The gel sieves the DNA by the size of the DNA molecule whereby smaller molecules travel faster. Double-stranded DNA moves at a rate that is approximately inversely proportional to the logarithm of the number of base pairs. This relationship however breaks down with very large DNA fragments and it is not possible to separate them using standard agarose gel electrophoresis. The limit of resolution depends on gel composition and field strength.[3] and the mobility of larger circular DNA may be more strongly affected than linear DNA by the pore size of the gel.[4] Separation of very large DNA fragments requires pulse field gel electrophoresis (PFGE). In field inversion gel electrophoresis (FIGE, a kind of PFGE), it is possible to have "band inversion" - where large molecules may move faster than small molecules.
Conformation of DNA
The conformation of the DNA molecule can significantly affect the movement of the DNA, for example, supercoiled DNA usually moves faster than relaxed DNA because it is tightly coiled and hence more compact. In a normal plasmid DNA preparation, multiple forms of DNA may be present,[5] and gel from the electrophoresis of the plasmids would normally show a main band which would be the negatively supercoiled form, while other forms of DNA may appear as minor fainter bands. These minor bands may be nicked DNA (open circular form) and the relaxed closed circular form which normally run slower than supercoiled DNA, and the single-stranded form (which can sometimes appear depending on the preparation methods) may move ahead of the supercoiled DNA. The rate at which the various forms move however can change using different electrophoresis conditions, for example linear DNA may run faster or slower than supercoiled DNA depending on conditions,[6] and the mobility of larger circular DNA may be more strongly affected than linear DNA by the pore size of the gel.[4] Unless supercoiled DNA markers are used, the size of a circular DNA like plasmid therefore may be more accurately gauged after it has been linearized by restriction digest.
DNA damage due to increased cross-linking will also reduce electrophoretic DNA migration in a dose-dependent way.[7][8]
Concentration of ethidium bromide
Circular DNA are more strongly affected by ethidium bromide concentration than linear DNA if ethidium bromide is present in the gel during electrophoresis. All naturally occurring DNA circles are underwound, but ethidium bromide which intercalates into circular DNA can change the charge, length, as well as the superhelicity of the DNA molecule, therefore its presence during electrophoresis can affect its movement in gel. Increasing ethidium bromide intercalated into the DNA can change it from a negatively supercoiled molecule into a fully relaxed form, then to positively coiled superhelix at maximum intercalation.[9] Agarose gel electrophoresis can be used to resolve circular DNA with different supercoiling topology.
Gel concentration
The concentration of the gel determines the pore size of the gel which affect the migration of DNA. The resolution of the DNA changes with the percentage concentration of the gel. Increasing the agarose concentration of a gel reduces the migration speed and improves separation of smaller DNA molecules, while lowering gel concentration permits large DNA molecules to be separated. For a standard agarose gel electrophoresis, a 0.7% gives good separation or resolution of large 5–10kb DNA fragments, while 2% gel gives good resolution for small 0.2–1kb fragments. Up to 3% can be used for separating very tiny fragments but a vertical polyacrylamide gel would be more appropriate for resolving small fragments. High concentrations gel however requires longer run times (sometimes days) and high percentage gels are often brittle and may not set evenly. High percentage agarose gels should be run with PFGE or FIGE. Low percentage gels (0.1−0.2%) are fragile and may break. 1% gels are common for many applications.[10]
Applied field
At low voltages, the rate of migration of the DNA is proportional to the voltage applied, i.e. the higher the voltage, the faster the DNA moves. However, in increasing electric field strength, the mobility of high-molecular-weight DNA fragments increases differentially, and the effective range of separation decreases and resolution therefore is lower at high voltage. For optimal resolution of DNA greater than 2kb in size in standard gel electrophoresis, 5 to 8 V/cm is recommended.[6] Voltage is also limited by the fact that it heats the gel and may cause the gel to melt if a gel is run at high voltage for a prolonged period, particularly for low-melting point agarose gel.
The mobility of DNA however may change in an unsteady field. In a field that is periodically reversed, the mobility of DNA of a particular size may drop significantly at a particular cycling frequency.[11] This phenomenon can result in band inversion whereby larger DNA fragments move faster than smaller ones in PFGE.
Mechanism of migration and separation
The negative charge of its phosphate backbone moves the DNA towards the positively charged anode during electrophoresis. However, the migration of DNA molecules in solution, in the absence of a gel matrix, is independent of molecular weight during electrophoresis, i.e. there is no separation by size without a gel matrix.[12] Hydrodynamic interaction between different parts of the DNA are cut off by streaming counterions moving in the opposite direction, so no mechanism exists to generate a dependence of velocity on length on a scale larger than screening length of about 10 nm.[11] This makes it different from other processes such as sedimentation or diffusion where long-ranged hydrodynamic interaction are important.
The gel matrix is therefore responsible for the separation of DNA by size during electrophoresis, however the precise mechanism responsible the separation is not entirely clear. A number of models exists for the mechanism of separation of biomolecules in gel matrix, a widely accepted one is the Ogston model which treats the polymer matrix as a sieve consisting of randomly distributed network of inter-connected pores.[13] A globular protein or a random coil DNA moves through the connected pores large enough to accommodate its passage, and the movement of larger molecules is more likely to be impeded and slowed down by collisions with the gel matrix, and the molecules of different sizes can therefore be separated in this process of sieving.[11]
The Ogston model however breaks down for large molecules whereby the pores are significantly smaller than size of the molecule. For DNA molecules of size greater than 1 kb, a reptation model (or its variants) is most commonly used. This model assumes that the DNA can crawl in a "snake-like" fashion (hence "reptation") through the pores as an elongated molecule. At higher electric field strength, this turned into a biased reptation model, whereby the leading end of the molecule become strongly biased in the forward direction, and this leading edge pulls the rest of the molecule along. In the fully biased mode, the mobility reached a saturation point and DNA beyond a certain size cannot be separated.[13] Perfect parallel alignment of the chain with the field however is not observed in practice as that would mean the same mobility for long and short molecules.[11] Further refinement of the biased reptation model takes into account of the internal fluctuations of the chain.[14]
The biased reptation model has also been used to explain the mobility of DNA in PFGE. The orientation of the DNA is progressively built up by reptation after the onset of a field, and the time it reached the steady state velocity is dependent on the size of the molecule. When the field is changed, larger molecules take longer to reorientate, it is therefore possible to discriminate between the long chains that cannot reach its steady state velocity from the short ones that travel most of the time in steady velocity.[14] Other models, however, also exist.
Real-time fluorescence microscopy of stained molecules showed more subtle dynamics during electrophoresis, with the DNA showing considerable elasticity as it alternately stretching in the direction of the applied field and then contracting into a ball, or becoming hooked into a U-shape when it gets caught on the polymer fibres.[15][16] This observation may be termed the "caterpillar" model.[17] Other model proposes that the DNA gets entangled with the polymer matrix, and the larger the molecule, the more likely it is to become entangled and its movement impeded.[18]
Visualization
The most common dye used to make DNA or RNA bands visible for agarose gel electrophoresis is ethidium bromide, usually abbreviated as EtBr. It fluoresces under UV light when intercalated into the major groove of DNA (or RNA). By running DNA through an EtBr-treated gel and visualizing it with UV light, any band containing more than ~20 ng DNA becomes distinctly visible. EtBr is a known mutagen, and safer alternatives are available, such as GelRed, produced by Biotium, which binds to the minor groove.[19]
SYBR Green I is another dsDNA stain, produced by Invitrogen. It is more expensive, but 25 times more sensitive, and possibly safer than EtBr, though there is no data addressing its mutagenicity or toxicity in humans.[20]
SYBR Safe is a variant of SYBR Green that has been shown to have low enough levels of mutagenicity and toxicity to be deemed nonhazardous waste under U.S. Federal regulations.[21] It has similar sensitivity levels to EtBr,[21] but, like SYBR Green, is significantly more expensive. In countries where safe disposal of hazardous waste is mandatory, the costs of EtBr disposal can easily outstrip the initial price difference, however.
Since EtBr stained DNA is not visible in natural light, scientists mix DNA with negatively charged loading buffers before adding the mixture to the gel. Loading buffers are useful because they are visible in natural light (as opposed to UV light for EtBr stained DNA), and they co-sediment with DNA (meaning they move at the same speed as DNA of a certain length). Xylene cyanol and Bromophenol blue are common dyes found in loading buffers; they run about the same speed as DNA fragments that are 5000 bp and 300 bp in length respectively, but the precise position varies with percentage of the gel. Other less frequently used progress markers are Cresol Red and Orange G which run at about 125 bp and 50 bp, respectively.
Visualization can also be achieved by transferring DNA after SDS-PAGE to a nitrocellulose membrane followed by exposure to a hybridization probe. This process is termed Southern blotting.
For fluorescent dyes, after electrophoresis the gel is illuminated with an ultraviolet lamp (usually by placing it on a light box, while using protective gear to limit exposure to ultraviolet radiation). The illuminator apparatus mostly also contains imaging apparatus that takes an image of the gel, after illumination with UV radiation. The ethidium bromide fluoresces reddish-orange in the presence of DNA, since it has intercalated with the DNA. The DNA band can also be cut out of the gel, and can then be dissolved to retrieve the purified DNA. The gel can then be photographed usually with a digital or polaroid camera. Although the stained nucleic acid fluoresces reddish-orange, images are usually shown in black and white (see figures). UV damage to the DNA sample can reduce the efficiency of subsequent manipulation of the sample, such as ligation and cloning. Shorter wavelength UV radiations (302 or 312 nm) cause greater damage, for example exposure for as little as 45 seconds can significantly reduce transformation efficiency. Therefore if the DNA is to be use for downstream procedures, exposure to a shorter wavelength UV radiations should be limited, instead higher-wavelength UV radiation (365 nm) which cause less damage should be used. Higher wavelength radiations however produces weaker fluorescence, therefore if it is necessary to capture the gel image, a shorter wavelength UV light can be used a short time. Addition of Cytidine or guanosine to the electrophoresis buffer at 1 mM concentration may protect the DNA from damage.[22] Alternatively, a blue light excitation source with a blue-excitable stain such as SYBR Green or GelGreen may be used.
Gel electrophoresis research often takes advantage of software-based image analysis tools, such as ImageJ.
| 1 | 2 | 3 |
|---|---|---|
 A 1% agarose 'slab' gel under normal light, behind a perspex UV shield. Only the marker dyes can be seen |
 The gel with UV illumination, the ethidium bromide stained DNA glows orange |
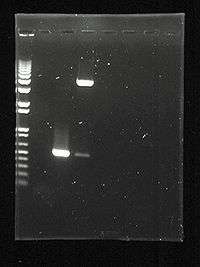 Digital photo of the gel. Lane 1. Commercial DNA Markers (1kbplus), Lane 2. empty, Lane 3. a PCR product of just over 500 bases, Lane 4. Restriction digest showing a similar fragment cut from a 4.5 kb plasmid vector |
References
- Jaguva Vasudevan, Ananda Ayyappan; Mario Perkovic; Yannick Bulliard; Klaus Cichutek; Didier Trono; Dieter Häussinger; Carsten Münk (August 2013). "Prototype Foamy Virus Bet Impairs the Dimerization and Cytosolic Solubility of Human APOBEC3G". Journal of Virology. 87 (16): 9030–9040. doi:10.1128/JVI.03385-12. PMC 3754047. PMID 23760237.
- G. Lucotte; F. Baneyx (1993). Introduction to Molecular Cloning Techniques. Wiley-Blackwell. p. 32. ISBN 978-0471188490.
- Joseph Sambrook; David Russell. "Chapter 5, protocol 1". Molecular Cloning - A Laboratory Manual. 1 (3rd ed.). p. 5.2. ISBN 978-0-87969-577-4.
- Aaij C, Borst P (1972). "The gel electrophoresis of DNA". Biochim Biophys Acta. 269 (2): 192–200. doi:10.1016/0005-2787(72)90426-1. PMID 5063906.
- Richard R. Sinden (24 November 1994). DNA Structure and Function. Academic Press Inc. p. 97. ISBN 978-0126457506.
- Joseph Sambrook; David Russell. "Chapter 5, protocol 1". Molecular Cloning - A Laboratory Manual. 1 (3rd ed.). pp. 5.5–5.6. ISBN 978-0-87969-577-4.
- Blasiak J, Trzeciak A, Malecka-Panas E, Drzewoski J, Wojewódzka M (2000). "In vitro genotoxicity of ethanol and acetaldehyde in human lymphocytes and the gastrointestinal tract mucosa cells". Toxicology in Vitro. 14 (4): 287–295. doi:10.1016/S0887-2333(00)00022-9. PMID 10906435.
- Lu Y, Morimoto K (2009). "Is habitual alcohol drinking associated with reduced electrophoretic DNA migration in peripheral blood leukocytes from ALDH2-deficient male Japanese?". Mutagenesis. 24 (4): 303–308. doi:10.1093/mutage/gep008. PMID 19286920.
- Donald Voet; Judith G. Voet (1995). Biochemistry (2nd ed.). John Wiley & Sons. pp. 877–878. ISBN 978-0471586517.
- "Agarose gel electrophoresis (basic method)". Biological Protocols. Retrieved 23 August 2011.
- Zimm BH, Levene SD (1992). "Problems and prospects in the theory of gel electrophoresis of DNA" (PDF). Quarterly Reviews of Biophysics. 25 (2): 171–204. doi:10.1017/s0033583500004662. PMID 1518924.
- Robert W. Old; Sandy B. Primrose (27 September 1994). Principle of Gene Manipulation - An Introduction to Genetic Engineering (5th ed.). Blackwell Scientific. p. 9. ISBN 9780632037124.
- Li Zhu; Hong Wang (2 March 2009). "Chapter 4 - Genetic Analysis in Miniaturized Electrophoresis Systems". In Tian, Wei-Cheng; Finehout, Erin (eds.). Microfluidics for Biological Applications. Springer. p. 125. ISBN 978-0-387-09480-9.
- Jean-Louis Viovy (2000). "Electrophoresis of DNA and other polyelectrolytes: Physical mechanisms". Reviews of Modern Physics. 72 (3): 813–872. Bibcode:2000RvMP...72..813V. doi:10.1103/RevModPhys.72.813.
- Smith SB, Aldridge PK, Callis JB (1989). "Observation of individual DNA molecules undergoing gel electrophoresis". Science. 243 (4888): 203–206. Bibcode:1989Sci...243..203S. doi:10.1126/science.2911733. PMID 2911733.
- Schwartz DC, Koval M (1989). "Conformational dynamics of individual DNA molecules during gel electrophoresis". Nature. 338 (6215): 520–2. Bibcode:1989Natur.338..520S. doi:10.1038/338520a0. PMID 2927511.
- David Sheehan (2009), Physical Biochemistry: Principles and Applications (2nd ed.), Wiley-Blackwell, p. 181, ISBN 978-0470856031
- Forster RE, Hert DG, Chiesl TN, Fredlake CP, Barron AE (2009). "DNA migration mechanism analyses for applications in capillary and microchip electrophoresis". Electrophoresis. 30 (12): 2014–24. doi:10.1002/elps.200900264. PMC 2762034. PMID 19582705.
- "SYBR Green I Nucleic Acid Gel Stain" (PDF). Archived from the original (PDF) on 2012-05-22. Retrieved 2013-06-23.
- "SYBR Safe DNA Gel Stain" (PDF). Archived from the original (PDF) on 2012-09-07. Retrieved 2013-06-23.
- Gründemann D, Schömig E. (1996). "Protection of DNA during preparative agarose gel electrophoresis against damage induced by ultraviolet light" (PDF). BioTechniques. 21 (5): 898–903. doi:10.2144/96215rr02. PMID 8922632. Archived from the original (PDF) on 2016-03-04. Retrieved 2017-11-26.