Advanced sleep phase disorder
Advanced Sleep Phase Disorder (ASPD), also known as the advanced sleep-phase type (ASPT) of circadian rhythm sleep disorder, is a condition that is characterized by a recurrent pattern of early evening (e.g. 7-9 pm) sleepiness and early morning awakening. This sleep phase advancement can interfere with daily social and work schedules, and results in shortened sleep duration and excessive daytime sleepiness.[1] The timing of sleep and melatonin levels are regulated by the body's central circadian clock, which is located in the suprachiasmatic nucleus in the hypothalamus.[2]
| Advanced Sleep Phase Disorder | |
|---|---|
| Specialty | Chronobiology |
| Symptoms | Earlier than desired onset and offset of sleep |
| Complications | Sleep deprivation |
| Risk factors | Increased incidence with age |
| Diagnostic method | Polysomnography, Horne-Ostberg morningness-eveningness questionnaire |
| Treatment | Bright light therapy, chronotherapy |
Symptoms
Individuals with ASPD report being unable to stay awake until conventional bedtime, falling asleep early in the evening, and being unable to stay asleep until their desired waking time, suffering early morning insomnia. When someone has advanced sleep phase disorder their melatonin levels and core body temperature cycle hours earlier than an average person.[3] These symptoms must be present and stable for a substantial period of time to be correctly diagnosed.
Diagnosis
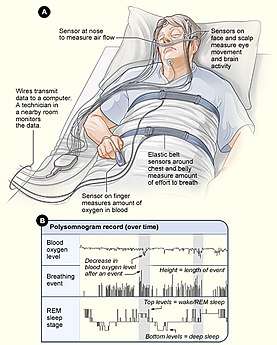
Individuals expressing the above symptoms may be diagnosed with ASPD using a variety of methods and tests. Sleep specialists measure the patient's sleep onset and offset, dim light melatonin onset, and evaluate Horne-Ostberg morningness-eveningness questionnaire results. Sleep specialists may also conduct a polysomnography test to rule out other sleep disorders like narcolepsy. Age and family history of the patient is also taken into consideration.[2]
Treatment
Once diagnosed, ASPD may be treated with bright light therapy in the evenings, or behaviorally with chronotherapy, in order to delay sleep onset and offset. The use of pharmacological approaches to treatment are less successful due to the risks of administering sleep-promoting agents early in the morning.[1] Additional methods of treatment, like timed melatonin administration or hypnotics have been proposed, but determining their safety and efficacy will require further research.[4] Unlike other sleep disorders, ASPD does not necessarily disrupt normal functioning at work during the day and some patients may not complain of excessive daytime sleepiness. Social obligations may cause an individual to stay up later than their circadian rhythm requires, however, they will still wake up very early. If this cycle continues, it can lead to chronic sleep deprivation and other sleep disorders.[3]
Epidemiology
ASPD is more common among middle and older adults. The estimated prevalence of ASPD is about 1% in middle-age adults, and is believed to affect men and women equally. The disorder has a strong familial tendency, with 40-50% of affected individuals having relatives with ASPD.[5] A genetic basis has been demonstrated in one form of ASPD, familial advanced sleep phase disorder (FASPS), which implicates missense mutations in genes hPER2 and CKIdelta in producing the advanced sleep phase phenotype.[5] The identification of two different genetic mutations suggests that there is heterogeneity of this disorder.[1]
Familial advanced sleep phase syndrome
FASPS Symptoms
While advanced sleep and wake times are relatively common, especially among older adults, the extreme phase advance characteristic of familial advanced sleep phase syndrome (also known as familial advanced sleep phase disorder) is rare. Individuals with FASPS fall asleep and wake up 4–6 hours earlier than the average population, generally sleeping from 7:30pm to 4:30am. They also have a free running circadian period of 22 hours, which is significantly shorter than the average human period of slightly over 24 hours.[6] The shortened period associated with FASPS results in a shortened period of activity, causing earlier sleep onset and offset. This means that individuals with FASPS must delay their sleep onset and offset each day in order to entrain to the 24-hour day. On holidays and weekends, when the average person's sleep phase is delayed relative to their workday sleep phase, individuals with FASPS experience further advance in their sleep phase.[7]
Aside from the unusual timing of sleep, FASPS patients experience normal quality and quantity of sleep. Like general ASPD, this syndrome does not inherently cause negative impacts, however, sleep deprivation may be imposed by social norms causing individuals to delay sleep until a more socially acceptable time, causing them to losing sleep due to earlier-than-usual wakeup time.[7]
Another factor that distinguishes FASPS from other advanced sleep phase disorders is its strong familial tendency and life-long expression. Studies of affected lineages have found that approximately 50% of directly-related family members experience the symptoms of FASPS, which is an autosomal dominant trait.[8] Diagnosis of FASPS can be confirmed through genetic sequencing analysis by locating genetic mutations known to cause the disorder. Treatment with sleep and wake scheduling and bright light therapy can be used to try to delay sleep phase to a more conventional time frame, however treatment of FASPS has proven largely unsuccessful.[9] Bright light exposure in the evening (between 7:00 and 9:00), during the delay zone as indicated by the phase response curve to light,[5] has been shown to delay circadian rhythms, resulting in later sleep onset and offset in patients with FASPS or other advanced sleep phase disorders.[1]
Discovery
In 1999, Louis Ptáček conducted a study at the University of Utah in which he coined the term familial advanced sleep phase disorder after identifying individuals with a genetic basis for an advanced sleep phase. The first patient evaluated during the study reported "disabling early evening sleepiness" and "early morning awakening"; similar symptoms were also reported in her family members. Consenting relatives of the initial patient were evaluated, as well as those from two additional families. The clinical histories, sleep logs and actigraphy patterns of subject families were used to define a hereditary circadian rhythm variant associated with a short endogenous (i.e. internally-derived) period. The subjects demonstrated a phase advance of sleep-wake rhythms that was distinct not only from control subjects, but also to sleep-wake schedules widely considered to be conventional. The subjects were also evaluated using the Horne-Östberg questionnaire, a structured self-assessment questionnaire used to determine morningness-eveningness in human circadian rhythms. The Horne-Östberg scores of first-degree relatives of affected individuals were higher than those of 'marry-in' spouses and unrelated control subjects. While much of morning and evening preference is heritable, the allele causing FASPS was hypothesized to have a quantitatively larger effect on clock function than the more common genetic variations that influence these preferences. Additionally, the circadian phase of subjects was determined using plasma melatonin and body core temperature measurements; these rhythms were both phase-advanced by 3–4 hours in FASPS subjects compared with control subjects. The Ptáček group also constructed a pedigree of the three FASPS kindreds which indicated a clear autosomal dominant transmission of the sleep phase advance.[10]
In 2001, the research group of Phyllis C. Zee phenotypically characterized an additional family affected with ASPS. This study involved an analysis of sleep/wake patterns, diurnal preferences (using a Horne-Östberg questionnaire), and the construction of a pedigree for the affected family. Consistent with established ASPS criteria, the evaluation of subject sleep architecture indicated that the advanced sleep phase was due to an alteration of circadian timing rather than an exogenous (i.e. externally-derived) disruption of sleep homeostasis, a mechanism of sleep regulation. Furthermore, the identified family was one in which an ASPS-affected member was present in every generation; consistent with earlier work done by the Ptáček group, this pattern suggests that the phenotype segregates as a single gene with an autosomal dominant mode of inheritance.[11]
In 2001, the research groups of Ptáček and Ying-Hui Fu published a genetic analysis of subjects experiencing the advanced sleep phase, implicating a mutation in the CK1-binding region of PER2 in producing the FASPS behavioral phenotype.[12] FASPS is the first disorder to link known core clock genes directly with human circadian sleep disorders.[13] As the PER2 mutation is not exclusively responsible for causing FASPS, current research has continued to evaluate cases in order to identify new mutations that contribute to the disorder.
Mechanisms (Per2 and CK1)
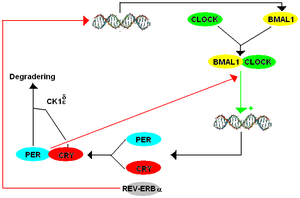
Two years after reporting the finding of FASPS, Ptáček's and Fu's groups published results of genetic sequencing analysis on a family with FASPS. They genetically mapped the FASPS locus to chromosome 2q where very little human genome sequencing was then available. Thus, they identified and sequenced all the genes in the critical interval. One of these was Period2 (Per2) which is a mammalian gene sufficient for the maintenance of circadian rhythms. Sequencing of the hPer2 gene ('h' denoting a human strain, as opposed to Drosophila or mouse strains) revealed a serine-to-glycine point mutation in the Casein Kinase I (CK1) binding domain of the hPER2 protein that resulted in hypophosphorylation of hPER2 in vitro.[12] The hypophosphorylation of hPER2 disrupts the transcription-translation (negative) feedback loop (TTFL) required for regulating the stable production of hPER2 protein. In a wildtype individual, Per2 mRNA is transcribed and translated to form a PER2 protein. Large concentrations of PER2 protein inhibits further transcription of Per2 mRNA. CK1 regulates PER2 levels by binding to a CK1 binding site on the protein, allowing for phosphorylation which marks the protein for degradation, reducing protein levels. Once proteins become phosphorylated, PER2 levels decrease again, and Per2 mRNA transcription can resume. This negative feedback regulates the levels and expression of these circadian clock components.
Without proper phosphorylation of hPER2 in the instance of a mutation in the CK1 binding site, less Per2 mRNA is transcribed and the period is shortened to less than 24 hours. Individuals with a shortened period due to this phosphorylation disruption entrain to a 24h light-dark cycle, which may lead to a phase advance, causing earlier sleep and wake patterns. However, a 22h period does not necessitate a phase shift, but a shift can be predicted depending on the time the subject is exposed to the stimulus, visualized on a Phase Response Curve (PRC).[14] This is consistent with studies of the role of CK1ɛ (a unique member of the CK1 family)[15] in the TTFL in mammals and more studies have been conducted looking at specific regions of the Per2 transcript.[16][17] In 2005, Fu's and Ptáček's labs reported discovery of a mutation in CKIδ (a functionally redundant form of CK1ɛ in the phosphorylation process of PER2) also causing FASPS. An A-to-G missense mutation resulted in a threonine-to-alanine alteration in the protein.[18] This mutation prevented the proper phosphorylation of PER2. The evidence for both a mutation in the binding domain of PER2 and a mutation in CKIδ as causes of FASPS is strengthened by the lack of the FASPS phenotype in wild type individuals and by the observed change in the circadian phenotype of these mutant individuals in vitro and an absence of said mutations in all tested control subjects. Fruit flies and mice engineered to carry the human mutation also demonstrated abnormal circadian phenotypes, although the mutant flies had a long circadian period while the mutant mice had a shorter period.[19][12] The genetic differences between flies and mammals that account for this difference circadian phenotypes are not known. Most recently, Ptáček and Fu reported additional studies of the human Per2 S662G mutation and generation of mice carrying the human mutation. These mice had a circadian period almost 2 hours shorter than wild-type animals under constant darkness. Genetic dosage studies of CKIδ on the Per2 S662G mutation revealed that depending on the binding site on Per2 that CK1δ interacts with, CK1δ may lead to hypo- or hyperphosphorylation of the Per2 gene.[20]
References
- Dodson, Ehren R.; Zee, Phyllis C (2010). "Therapeutics for Circadian Rhythm Sleep Disorders". Sleep Medicine Clinics. 5 (4): 701–715. doi:10.1016/j.jsmc.2010.08.001. ISSN 1556-407X. PMC 3020104. PMID 21243069.
- Reid KJ, Chang AM, Dubocovich ML, Turek FW, Takahashi JS, Zee PC (July 2001). "Familial advanced sleep phase syndrome". Archives of Neurology. 58 (7): 1089–94. doi:10.1001/archneur.58.7.1089. PMID 11448298.
- "Advanced Sleep-Wake Phase Disorder - Symptoms, Diagnosis, Treatment".
- Zhdanova, Irina V.; Vitiello, Michael V.; Wright, Kenneth P.; Carskadon, Mary A.; Auger, R. Robert; Auckley, Dennis; Sack, Robert L. (1 November 2007). "Circadian Rhythm Sleep Disorders: Part II, Advanced Sleep Phase Disorder, Delayed Sleep Phase Disorder, Free-Running Disorder, and Irregular Sleep-Wake Rhythm". Sleep. 30 (11): 1484–1501. doi:10.1093/sleep/30.11.1484. ISSN 0161-8105. PMC 2082099. PMID 18041481.
- Zhu, Lirong; Zee, Phyllis C. (2012). "Circadian Rhythm Sleep Disorders". Neurologic Clinics. 30 (4): 1167–1191. doi:10.1016/j.ncl.2012.08.011. ISSN 0733-8619. PMC 3523094. PMID 23099133.
- Jones, Christopher R.; Huang, Angela L.; Ptáček, Louis J.; Fu, Ying-Hui (2013). "Genetic Basis of Human Circadian Rhythm Disorders". Experimental Neurology. 243: 28–33. doi:10.1016/j.expneurol.2012.07.012. ISSN 0014-4886. PMC 3514403. PMID 22849821.
- Tafti, Mehdi; Dauvilliers, Yves; Overeem, Sebastiaan (2007). "Narcolepsy and familial advanced sleep-phase syndrome: molecular genetics of sleep disorders". Current Opinion in Genetics & Development. 17 (3): 222–227. doi:10.1016/j.gde.2007.04.007. PMID 17467264.
- Pack, Allan I.; Pien, Grace W. (18 February 2011). "Update on Sleep and Its Disorders". Annual Review of Medicine. 62 (1): 447–460. doi:10.1146/annurev-med-050409-104056. ISSN 0066-4219. PMID 21073334.
- Fu, Y. H.; Ptáček, L. J.; Chong, S. Y. (2012). "Genetic insights on sleep schedules: this time, it's PERsonal". Trends in Genetics. 28 (12): 598–605. doi:10.1016/j.tig.2012.08.002. ISSN 0168-9525. PMC 3500418. PMID 22939700.
- Jones, Christopher R.; Campbell, Scott S.; Zone, Stephanie E.; Cooper, Fred; DeSano, Alison; Murphy, Patricia J.; Jones, Bryan; Czajkowski, Laura; Ptček, Louis J. (1999). "Familial advanced sleep-phase syndrome: A short-period circadian rhythm variant in humans". Nature Medicine. 5 (9): 1062–1065. doi:10.1038/12502. ISSN 1078-8956. PMID 10470086.
- Reid, Kathryn J.; Chang, Anne-Marie; Dubocovich, Margarita L.; Turek, Fred W.; Takahashi, Joseph S.; Zee, Phyllis C. (1 July 2001). "Familial Advanced Sleep Phase Syndrome". Archives of Neurology. 58 (7): 1089–94. doi:10.1001/archneur.58.7.1089. ISSN 0003-9942. PMID 11448298.
- Toh, K. L. (9 February 2001). "An hPer2 Phosphorylation Site Mutation in Familial Advanced Sleep Phase Syndrome". Science. 291 (5506): 1040–1043. Bibcode:2001Sci...291.1040T. doi:10.1126/science.1057499. PMID 11232563.
- Takahashi, Joseph S.; Hong, Hee-Kyung; Ko, Caroline H.; McDearmon, Erin L. (2008). "The genetics of mammalian circadian order and disorder: implications for physiology and disease". Nature Reviews Genetics. 9 (10): 764–775. doi:10.1038/nrg2430. ISSN 1471-0056. PMC 3758473. PMID 18802415.
- Johnson, Carl H. (2013). "Entrainment of Circadian Programs" (PDF). Chronobiology International. 20 (5): 741–774. doi:10.1081/CBI-120024211. PMID 14535352.
- Yang, Yu; Xu, Tingting; Zhang, Yunfei; Qin, Ximing (2017). "Molecular basis for the regulation of the circadian clock kinases CK1δ and CK1ε". Cellular Signalling. 31: 58–65. doi:10.1016/j.cellsig.2016.12.010. ISSN 0898-6568. PMID 28057520.
- Vanselow, Katja; Vanselow, Jens T.; Westermark, Pål O.; Reischl, Silke; Maier, Bert; Korte, Thomas; Herrmann, Andreas; Herzel, Hanspeter; Schlosser, Andreas (1 October 2006). "Differential effects of PER2 phosphorylation: molecular basis for the human familial advanced sleep phase syndrome (FASPS)". Genes & Development. 20 (19): 2660–2672. doi:10.1101/gad.397006. ISSN 0890-9369. PMC 1578693. PMID 16983144.
- Menaker, M.; Ralph, M. R. (2 September 1988). "A mutation of the circadian system in golden hamsters". Science. 241 (4870): 1225–1227. Bibcode:1988Sci...241.1225R. doi:10.1126/science.3413487. ISSN 0036-8075. PMID 3413487.
- Xu, Ying; Kong L. Toh; Christopher R. Jones; et al. (12 January 2007). "Modeling of a human circadian mutation yields insights into clock regulation by PER2". Cell. 128 (1): 59–70. doi:10.1016/j.cell.2006.11.043. PMC 1828903. PMID 17218255.
- Xu, Ying; Quasar S. Padiath; Robert E. Shapiro; et al. (31 March 2005). "Functional consequences of a CKIδ mutation causing familial advanced sleep phase syndrome". Nature. 434 (7033): 640–644. Bibcode:2005Natur.434..640X. doi:10.1038/nature03453. PMID 15800623.
- Xu, Y.; Toh, K.L.; Jones, C.R.; Shin, J.-Y.; Fu, Y.-H.; Ptáček, L.J. (2007). "Modeling of a Human Circadian Mutation Yields Insights into Clock Regulation by PER2". Cell. 128 (1): 59–70. doi:10.1016/j.cell.2006.11.043. PMC 1828903. PMID 17218255.