FANTOM
FANTOM (Functional Annotation of the Mouse/Mammalian Genome) is an international research consortium first established in 2000 as part of the RIKEN research institute in Japan.[1] The original meeting gathered international scientists from diverse backgrounds to help annotate the function of mouse cDNA clones generated by the Hayashizaki group.[2] Since the initial FANTOM1 effort, the consortium has released multiple projects that look to understand the mechanisms governing the regulation of mammalian genomes.[1] Their work has generated a large collection of shared data and helped advance biochemical and bioinformatic methodologies in genomics research.
Foundation
In 1995, researchers of the RIKEN institute began creating an encyclopedia of full length cDNAs for the mouse genome. The goal of this 'Mouse Encyclopedia Project' was to provide a functional annotation of the mouse transcriptome. This mapping would provide a valuable resource for gene discovery, understanding disease-causing genes and homology across species. This promised to be a formidable task from the onset. Current methodologies were insufficient to generate full length cDNA clones at scale, and to be useful as a resource the annotations would have to be agreed upon by experts across different disciplines.[1][2]
The first goal was to develop methods that allowed generation of full length cDNA libraries. Reverse transcriptase protocols at the time had difficulties with the secondary structure of mRNA, leading to abbreviated cDNAs that were difficult to align and invited further complications in downstream analysis. To surpass this limitation, a method utilizing trehalose was developed to allow reverse transcriptase to function at a higher temperature, relaxing secondary structures.[3] Other methods were additionally developed to assist in the construction of clonal cDNA libraries. These include a biotin-based capture system to select for full length cDNA, a novel lambda phage vector that minimized biases when delivering cDNA into a plasmid, and an iterative strategy to enrich for cDNA that had yet to be sequenced.[1][2][4][5][6]
Sequencing began in 1998 and progressed rapidly, producing 246 cDNA libraries that encompassed 21,076 cDNA clones across a large range of mouse cells and tissues. While this stage was largely successful, further limitations were encountered at the bioinformatic level. The sequenced cDNAs were annotated in a semi-automatic manner that utilized available databases (such as species homology and known protein motifs) to assign genes within a Gene Ontology (GO) framework. However, many novel sequences did not have meaningful matches when BLAST against gene databases.[1][2]
After consulting Gerry Rubin, the organizer of the first genome annotation effort for Drosophila melanogaster, it became apparent that a robust system for annotation that incorporated computational prediction and manual curation was required for the novel sequences. Desiring input from experts in bioinformatics, genetics and other scientific fields, the RIKEN group organized the first FANTOM meeting.
FANTOM1
To facilitate the annotation of the mouse cDNA clones, the RIKEN research group developed a web-based service called FANTOM+ prior to the first meeting. Users could search for motifs, view pre-computed sequence similarity scores, as well as query other public databases and integrate relevant annotations into the FANTOM database. The assignment and functional annotation of the genes required multiple bioinformatic tools and databases. Predominant tools included BLASTN/BLASTX, FASTA/FASTY, DECODER, EST-WISE and HMMER, while both nucleic acid and protein databases such as SwissProt, UniGene and NCBI-nr were utilized. Concurrently, a collaboration with the Mouse Genome Informatics group (MGI) allowed the RIKEN researchers to establish a validated set of clones that were identical between the two databases.[1][2]
Armed with computational methodologies and over 20,000 cDNA sequences, the RIKEN group organized the first FANTOM meeting in Tsukuba City from August 28 to September 8, 2000. A diverse group of international scientists were recruited to discuss strategies and execute the annotation of the RIKEN clones. The assembled computational procedures allowed for sequence comparison and domain analysis to assign putative function using GO terms. Redundancy of the cDNA clones presented a challenge, requiring clustering strategies and referral to the MGI validation set to identify unique clones. The RIKEN set of clones was eventually reduced to 15,295 genes, although this was cautiously considered an overestimation.[1][2]
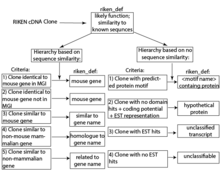
Central to the curation efforts was the creation of the RIKEN definition. This provided a hierarchical and systematic means to assign functions to the clones based upon known genes, placing priority on previously established or well-curated knowledge. The hierarchical nature of the classification allowed for consistency when a sequence was highly similar to multiple different genes. Importantly, if no sequence similarity was found, the definition assigned putative function based upon predicted protein motif signatures, coding potential and matches to expressed sequence tag (EST) databases. Only in the absence of any predicted or representative similarity would a clone be considered ‘unclassifiable.’[1][2]
The collected efforts of RIKEN/FANTOM resulted in a 2001 Nature publication.[7] The results included the assignment of the 21,076 cDNA clones to 4,012 GO terms, identification of novel mouse genes and protein motifs, detection of likely alternative spliceforms, and the discovery of mouse genes orthologous to human disease genes. Additionally, the first sequenced human genome was published a week later and incorporated FANTOM’s results to predict the number of human genes.[1][2][8]
FANTOM2
Having established and improved upon the protocols for full-length cDNA library generation, the RIKEN group continued to add to the FANTOM collection. Modifications to their methods allowed for further selection of rare and long transcripts, enabling identification of cDNA over 4kb in length. The second FANTOM meeting occurred May 2002 - by then the number of cDNA clones had increased by 39,694 to a total of 60,770.[1][9]
One insight gained from FANTOM1 was that alternative polyadenylation was common in the mouse transcriptome, meaning that 3’-end clustering led to extensive redundancy. To address this, additional sequencing of the 5’-end was performed to identify unique clones. The FANTOM2 publication contributed a substantial addition of novel protein coding transcripts. Arguably the most notable result of FANTOM2 was that efforts to select for long and rare transcripts had revealed a significant amount of non protein-coding RNA.[1][9]
Again, the FANTOM collection proved to be a fruitful resource. The non-coding RNA were identified as antisense RNA and long non-coding RNAs (lncRNA), poorly understood classes of regulatory RNA.[10][11] The first published sequence of the mouse genome utilized the annotations established by FANTOM.[10] Other efforts were able to describe entire protein families, such as the G protein-coupled receptors.[1][12][13]
FANTOM3
An ultimate goal of FANTOM is to establish gene networks that capture the regulatory interactions of transcription, and to differentiate these interactions by cell type or state. To this extent, it was realized that the polymorphic nature of the 5'-end of sequences would require extensive mapping. Characterizing transcription start sites (TSSs) would allow identification of promoters and differentiation of their usage between cell types. This also meant further developments in sequencing methods were needed. While full length mouse cDNAs continued to be generated, the RIKEN-led researchers established Cap Analysis of Gene Expression (CAGE), a technique that would drive much of their future work.[1]
Development of CAGE
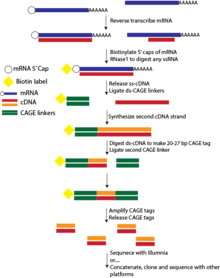
CAGE was a continuation of the concepts developed for FANTOM1 - and used extensively in the following projects - for capturing 5' mRNA caps. Unlike previous efforts to generate full length cDNA, CAGE examines fragments, or tags, that are 20-27 in length. This provided an economical and high-throughput means of mapping TSSs, including promoter structure and activity.[1]
The general steps are as follows: cDNA is reverse transcribed from mRNA using random or oligo dT primers. The cap trapper method is then employed to ensure selection of full length cDNA. This entails adding biotin to the 5' cap, and subsequent capture with streptavidin beads after an RNase digestion step to remove single stranded RNA that has not hybridized to cDNA. Following cap trapping, the cDNA is separated from the RNA-cDNA hybrid. A double-stranded CAGE linker that is also biotinylated is ligated to the 5' end of the cDNA, and the second strand of the cDNA is synthesized. This resulting dual stranded DNA is digested with the Mme1 endonuclease, cutting the CAGE linker and producing a 20-27bp CAGE tag. A second linker is added to the 3'-end and the tag is amplified by PCR. Finally, the CAGE tags are released from the 5' and 3' linkers. The tags can then be sequenced, concatenated or cloned.[4][14][15][16] At the time, CAGE was carried out using the RISA 384 capillary sequencer that had been previously established by RIKEN.[1]

Discoveries
The development of CAGE gave rise to a number of milestone findings. Importantly, RNA was found to be much more abundant in the mammalian transcriptome than previously thought, accompanied with the realization that the genome was pervasively transcribed.[1] Combining the methods of CAGE, gene identification signatures, and gene signature cloning, the ‘transcriptional landscape’ of the mammalian genome was mapped, characterizing the pattern of transcription control signals and the transcripts they generate.[17] It was discovered that there are many more transcripts than the estimated 22,000 genes in the mouse genome, and that many of these transcriptional units have alternative promoters and polyadenylation sites.
Furthermore, it was discovered that ‘transcriptional forests’, clusters of transcripts that share common expression regions and regulatory events, are separated by ‘transcription deserts,’ and make up ~63% of the genome.[17] A jointly released publication found that many of the transcripts in these forests show antisense transcription, and that most sense/antisense pairs show concordant regulation.[18] Another notable result showed that many non-coding RNAs are dynamically expressed, with many being initiated in 3’ untranslated regions, and that they are positionally conserved across species.[17]
The third milestone paper to come out of FANTOM3 investigated mammalian promoter architecture and evolution.[19] It established two classes of mammalian promoters. The first are TATA box-enriched promoters, with well defined transcriptional start sites. These promoters are evolutionary conserved and are more commonly associated with tissue-specific genes. The second and more common class of promoters, broad CpG rich promoters, are plastic, evolvable, and expressed in a wide range of cells and tissues. This study also demonstrated that CpG-rich promoters may be bidirectional (produce sense-antisense pairs), and are highly susceptible epigenetic control and are thus a potential component of adaptive evolution.
The meeting for FANTOM3 occurred in September, 2004. A collection of satellite publications that spawned from FANTOM3 were published in PLoS Genetics. They include further work on promoter properties, exon length and pseudo-messenger RNA.[20][21]
FANTOM4
The rise of next-generation sequencing was significantly beneficial to the advancement of CAGE technology. Using the Roche-454 sequencer, the FANTOM group developed deepCAGE, increasing the throughput of CAGE to more than a million tags per sample.[22] At these depths, researchers could now start constructing networks of gene regulatory interactions. The FANTOM4 meeting took place December, 2006.
While previous FANTOM projects examined a range of cell types, FANTOM4's purpose was to deeply interrogate the dynamics driving cellular differentiation. Analysis was confined to a human THP-1 cell line, providing time course data of a monoblast becoming a monocyte. DeepCage resolved TSSs at single-nucleotide resolution, pinpointing where transcription factors (TFs) bind. By monitoring time-dependent gene expression changes as cells differentiated, inference was provided for which regulatory motifs are predictive of expression changes, time dependency of TF activity, and TF target genes.[23] These efforts resulted in a transcriptional regulatory network, demonstrating that the differentiation process is highly complex and driven by a great magnitude of TFs enacting both positive and negative regulatory interactions.
FANTOM4 also increased our understanding of retrotransposon transcription and transcriptional initiation RNAs (tiRNAs). Retrotransposons contribute to repetitive elements in mammalian genomes and can affect multiple biological processes - like genomic evolution - as well as structures, such as alternative promoters and exons.[24][25] It was demonstrated that retrotransposons are expressed in a cell and tissue specific manner, and approximately 250,000 previously unknown retrotransposon-driven TSSs were identified.[26]
It was discovered that retrotransposons can influence mammalian transcription and transcriptional regulation of both coding and non-coding RNAs in various tissues.[26] Further efforts found a genomically and evolutionary widespread new class of RNAs, called transcription initiation RNAs (tiRNA).[27] This species of RNA are relatively tiny (~18 nucleotides long) and are typically found downstream of TSSs of CpG rich promoters. tiRNAs are low in abundance and are associated with highly expressed genes, as well as RNA polymerase II binding and TSSs. More recent work has shown that tiRNs may be able to modulate epigenetic states and local chromatin architecture.[28] However, it possible that these tiRNAs do not have a regulatory role and are simply a byproduct of transcription.[1][27]
Following these initial findings, an atlas of combinatorial transcriptional regulation in mouse and humans was published by the RIKEN researchers.[29] This work demonstrated that transcriptional complexes can interact within a network to control tissue identity/cell state, and that these networks are often dominated by ‘facilitator' transcription factors which are broadly expressed across tissues/cells. It was found that about half of the measured regulatory interactions were conserved between mouse and human. FANTOM4 led to numerous satellite papers, investigating topics like promoter architecture, miRNA regulation and genomic regulatory blocks.[30][31][32]
FANTOM5
The fifth round of FANTOM aimed to provide insight into the regulatory landscape of the transcriptome across as many cell states as possible.[1] It continues to be a relevant resource of shared data. The project consisted of two phases: the first focused on steady state cells, while the second focused on temporal data. Advancements in next generation sequencing were leveraged to achieve FANTOM5’s great breadth, with single molecule sequencing allowing single base pair resolution of TSS activity from as little as 100 ng of RNA.[33] Samples was collected from every human organ, as well as over 200 cancer lines, 30 time courses of cellular differentiation, mouse development time courses, and over 200 primary cell types. In total, 1,816 human and 1,1016 mouse samples were profiled across both phases.[33][34]
While similar to the ENCODE Project, FANTOM5 differs in two key ways. First, ENCODE utilized immortalised cell lines, while FANTOM5 focused on primary cells and tissues, which are more reflective of the actual biological processes responsible for maintaining cell type identity. Second, ENCODE utilized multiple genomic assays to capture the transcriptome and epigenome. FANTOM5 focused solely on the transcriptome, relying on other published work to infer features like cell type as defined by chromatin status.[1] The FANTOM5 meeting took place October, 2011.
Phase 1
The first phase of FANTOM5 involved taking ‘snapshots’ of a wide range of steady state cell types using CAGE profiling across 975 human and 399 mouse samples. This initial effort resulted in two Nature papers - one describing the mammalian promoter landscape and the other describing active enhancers.[35][36] Together, they provide an atlas of promoters, enhancers and TSSs across diverse cell types, acting as a ‘baseline’ for studying the complex landscape of transcription regulation. Specifically, single molecule CAGE profiles were generated using a HeliScope sequencer across 573 human primary cell samples, 128 mouse primary cell samples, 250 cancer cell lines, 152 human post-mortem tissues and 271 mouse developmental tissue samples.[33][37]
A new method to identify the CAGE peaks was developed, called decomposition peak analysis. CAGE tags are clustered by proximity, followed by independent component analysis to decompose the peaks into non-overlapping regions. An enrichment step is applied to ensure the peaks correspond to TSSs, and external data of EST, histone H3 lysine 4 trimethylation marks and DNase hypersensitivity sites are used to support that the peaks are genuine TSSs.[33]
A key finding showed that the typical mammalian promoter contains multiple TSSs with differing expression patterns across samples.[1][35] This implied that these TSSs are regulated separately, despite being within close proximity. Ubiquitously expressed promoters had the highest conservation in their sequences, while cell-specific promoters were less conserved. A further prominent result suggested that enhancer-derived RNA (eRNA) are transcribed in a cell/tissue specific manner, reflective of the activity of that enhancer.[37]
Phase 2
While the first phase was focused on a steady state representation of cell states, the second phase looked to explore the dynamic process of transitioning cell states through the use of time course data. Again, CAGE was employed - this time over 19 human and 14 mouse time courses covering a range of cell types and biological stimuli that represented 408 distinct time points. This included the differentiation of stem cell or committed progenitor cells towards their terminal fates, as well as fully differentiated cells responding to growth factors or pathogens.[1][33][38]
Unsupervised clustering was performed to identify a set of distinct response classes, examining patterns in expression fold changes compared to time 0. In this manner, the expression of enhancers, TF promoters and non-TF promoters were generalized on a temporal scale of the first 6 hours of the time-course. Generally, the earliest response of the cells occurred at enhancers, with eRNA concentrations peaking as early as 15 minutes after time 0. Even in the classes that represent ‘later’ responses, enhancers tended to activate before proximal promoters. Variability was seen in the persistence of this activation - some enhancers rapidly returned to baseline after the burst at 15 minutes, while others persisted after promoter activation. Together, this is suggestive that eRNA may have differential roles in regulating gene activity.[38]
Additional Work
Aside from the typical sharing of data on the FANTOM database, FANTOM5 also introduced two bioinformatic tools for data exploration. ZENBU is a genome browser with additional functionality: users can upload BAM files of CAGE, short-RNA and ChIP-seq experiments and perform quality control, normalization, peak finding and annotation among visual comparisons.[39] SSTAR (Semantic catalog of, samples, transcription initiations, and regulations) meanwhile allows exploration and searches of the FANTOM5 samples and their genomic features.[40]
The bounty of data produced by FANTOM5 continues to provide a resource for researchers looking to explain the regulatory mechanisms that shape processes like development. Often CAGE data in a specific cell/tissue type is used in conjunction with further epigenomic assays - one such example describes the interplay of DNA methylation and CAGE-defined regulatory sequences during differentiation of a granulocyte.[41]
Three years after introducing the enhancer and promoter atlases, the FANTOM group released atlases for lncRNAs and microRNAs (miRNA), incorporating FANTOM5 data.[42][43] An overarching goal was to provide further insight into the earlier observation of pervasive transcription of the mammalian genome. The lncRNA work characterized 27,919 human lncRNA genes across 1,829 samples to stimulate research in the functional relevance of this poorly understood class of RNA. The results were suggestive that 69% of the identified lncRNA had potential functionality, although more evidence is required to comment on whether the remaining 31% are merely transcriptional ‘noise’ from spurious transcription initiation. The miRNA atlas identified 1,357 human and 804 mouse miRNA promoters and demonstrated strong sequence conservation between the two species. It was also demonstrated that primary miRNA expression could be used as a proxy for mature miRNA levels.
FANTOM6
Currently underway, FANTOM6 aims to systematically characterize the role of lncRNA in the human genome. The biological function of these large (200+ nucleotides) and untranslated RNA is largely unknown. Based upon the few works that have examined lncRNA, it is believed that they are involved in regulating transcription, translation, post-translational modifications, and epigenetic marks. However, current knowledge of the extent and range of these putative regulatory interactions is rudimentary.[1][44]
There are numerous challenges to address for this next rendition of FANTOM. In particular, lncRNAs are ill-defined - they lack conservation and vary greatly in size, ranging from 200 to over one million nucleotides in length. Unlike coding transcripts, which are found in the cytosol for translation, lncRNA are found primarily in the nucleus - a much more complex landscape of RNA. In general, lncRNA have lower expression levels than coding transcripts, but there is great variability in this expression which can be obscured by cell type or localization within the nucleus. Furthermore, functional classification lncRNAs remains hotly debated - it is unknown if lncRNAs can be grouped based on common function/mechanisms of action, or by active domains.[1]
FANTOM has laid out a three pronged experimental strategy to explore these unknowns. A reference transcriptome and epigenome profile of different cell types will be constructed as a base line for each cell type. Next, using lncRNAs identified in previous publications, FANTOM5 data and further CAGE profiling, perturbation experiments will be conducted to evaluate changes in cellular molecular phenotype. Lastly, complementary technology will be used to functionally annotate/classify a selected subset of lncRNAs.[44] These techniques will be aimed at elucidating lncRNA secondary structure, their association to proteins and chromatin, and mapping long range interactions of lncRNA throughout the genome.[1]
References
- de Hoon, Michiel; Shin, Jay W.; Carninci, Piero (8 August 2015). "Paradigm shifts in genomics through the FANTOM projects". Mammalian Genome. 26 (9–10): 391–402. doi:10.1007/s00335-015-9593-8. PMC 4602071. PMID 26253466.
- "FANTOM introduction". fantom.gsc.riken.jp.
- Carninci, Piero; Nishiyama, Yoko; Westover, Arthur; et al. (20 January 1998). "Thermostabilization and thermoactivation of thermolabile enzymes by trehalose and its application for the synthesis of full length cDNA". Proceedings of the National Academy of Sciences of the United States of America. 95 (2): 520–524. doi:10.1073/pnas.95.2.520. ISSN 0027-8424. PMC 18452. PMID 9435224.
- Carninci, Piero; Kvam, Catrine; Kitamura, Akiko; et al. (November 1996). "High-Efficiency Full-Length cDNA Cloning by Biotinylated CAP Trapper". Genomics. 37 (3): 327–336. doi:10.1006/geno.1996.0567. PMID 8938445.
- Carninci, Piero; Shibata, Yuko; Hayatsu, Norihito; et al. (October 2000). "Normalization and Subtraction of Cap-Trapper-Selected cDNAs to Prepare Full-Length cDNA Libraries for Rapid Discovery of New Genes". Genome Research. 10 (10): 1617–1630. doi:10.1101/gr.145100. ISSN 1088-9051. PMC 310980. PMID 11042159.
- Carninci, Piero; Shibata, Yuko; Hayatsu, Norihito; et al. (September 2001). "Balanced-Size and Long-Size Cloning of Full-Length, Cap-Trapped cDNAs into Vectors of the Novel λ-FLC Family Allows Enhanced Gene Discovery Rate and Functional Analysis". Genomics. 77 (1–2): 79–90. doi:10.1006/geno.2001.6601. PMID 11543636.
- Kawai, J.; Shinagawa, A.; Shibata, K.; et al. (8 February 2001). "Functional annotation of a full-length mouse cDNA collection". Nature. 409 (6821): 685–690. doi:10.1038/35055500. PMID 11217851.
- Lander, Eric S.; Linton, Lauren M.; Birren, Bruce; Nusbaum, Chad; Zody, Michael C.; et al. (15 February 2001). "Initial sequencing and analysis of the human genome". Nature. 409 (6822): 860–921. doi:10.1038/35057062. PMID 11237011.
- Okazaki, Y.; Furuno, M.; Kasukawa, T.; Adachi, J.; Bono, H.; et al. (5 December 2002). "Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs". Nature. 420 (6915): 563–573. doi:10.1038/nature01266. PMID 12466851.
- Numata, K. (2 June 2003). "Identification of Putative Noncoding RNAs Among the RIKEN Mouse Full-Length cDNA Collection". Genome Research. 13 (6): 1301–1306. doi:10.1101/gr.1011603. PMC 403720. PMID 12819127.
- Kiyosawa, H. (2 June 2003). "Antisense Transcripts With FANTOM2 Clone Set and Their Implications for Gene Regulation". Genome Research. 13 (6): 1324–1334. doi:10.1101/gr.982903. PMC 403655. PMID 12819130.
- Okazaki, Y. (2 June 2003). "A Guide to the Mammalian Genome". Genome Research. 13 (6): 1267–1272. doi:10.1101/gr.1445603.
- Kawasawa, Y. (2 June 2003). "G Protein-Coupled Receptor Genes in the FANTOM2 Database". Genome Research. 13 (6): 1466–1477. doi:10.1101/gr.1087603. PMC 403690. PMID 12819145.
- "RIKEN CAGE technology". www.osc.riken.jp (in Japanese). Archived from the original on 2017-10-09. Retrieved 2018-02-27.
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T.; Fukuda, S.; Sasaki, D.; Podhajska, A.; Harbers, M.; Kawai, J.; Carninci, P.; Hayashizaki, Y. (8 December 2003). "Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage". Proceedings of the National Academy of Sciences. 100 (26): 15776–15781. doi:10.1073/pnas.2136655100. PMC 307644. PMID 14663149.
- Kodzius, Rimantas; Kojima, Miki; Nishiyori, Hiromi; Nakamura, Mari; Fukuda, Shiro; Tagami, Michihira; Sasaki, Daisuke; Imamura, Kengo; Kai, Chikatoshi; Harbers, Matthias; Hayashizaki, Yoshihide; Carninci, Piero (March 2006). "CAGE: cap analysis of gene expression". Nature Methods. 3 (3): 211–222. doi:10.1038/nmeth0306-211. PMID 16489339.
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M. C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; Kodzius, R.; Shimokawa, K.; Bajic, V. B.; Brenner, S. E.; Batalov, S.; Forrest, A. R.; Zavolan, M.; Davis, M. J.; Wilming, L. G.; Aidinis, V.; Allen, J. E.; Ambesi-Impiombato, A.; Apweiler, R.; Aturaliya, R. N.; Bailey, T. L.; Bansal, M.; Baxter, L.; Beisel, K. W.; Bersano, T.; et al. (2 September 2005). "The Transcriptional Landscape of the Mammalian Genome". Science. 309 (5740): 1559–1563. doi:10.1126/science.1112014. PMID 16141072.
- Katayama, S; Tomaru, Y; Kasukawa, T; et al. (2 September 2005). "Antisense Transcription in the Mammalian Transcriptome". Science. 309 (5740): 1564–1566. doi:10.1126/science.1112009. PMID 16141073.
- Carninci, Piero; Sandelin, Albin; Lenhard, Boris; Katayama, Shintaro; Shimokawa, Kazuro; et al. (2006). "Genome-wide analysis of mammalian promoter architecture and evolution". Nature Genetics. 38 (6): 626–635. doi:10.1038/ng1789. ISSN 1546-1718. PMID 16645617.
- "FANTOM3 - Papers". fantom.gsc.riken.jp.
- "PLOS Genetics: Search Results FANTOM3". journals.plos.org.
- de Hoon, Michiel; Hayashizaki, Yoshihide (April 2008). "Deep cap analysis gene expression (CAGE): genome-wide identification of promoters, quantification of their expression, and network inference". BioTechniques. 44 Supplement (4): 627–632. doi:10.2144/000112802. PMID 18474037.
- Suzuki, Harukazu; Consortium, The FANTOM; Forrest, Alistair R R; et al. (2009). "The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line". Nature Genetics. 41 (5): 553–562. doi:10.1038/ng.375. ISSN 1546-1718. PMC 6711855. PMID 19377474.
- Kazazian, H. H. (12 March 2004). "Mobile Elements: Drivers of Genome Evolution". Science. 303 (5664): 1626–1632. doi:10.1126/science.1089670. PMID 15016989.
- Finnegan, David J. (June 2012). "Retrotransposons". Current Biology. 22 (11): R432–R437. doi:10.1016/j.cub.2012.04.025. PMID 22677280.
- Faulkner, Geoffrey J; Kimura, Yasumasa; Daub, Carsten O; et al. (19 April 2009). "The regulated retrotransposon transcriptome of mammalian cells". Nature Genetics. 41 (5): 563–571. doi:10.1038/ng.368. PMID 19377475.
- Taft, Ryan J; Glazov, Evgeny A; Cloonan, Nicole; et al. (19 April 2009). "Tiny RNAs associated with transcription start sites in animals". Nature Genetics. 41 (5): 572–578. doi:10.1038/ng.312. PMID 19377478.
- Taft, Ryan J; Hawkins, Peter G; Mattick, John S; Morris, Kevin V (2011). "The relationship between transcription initiation RNAs and CCCTC-binding factor (CTCF) localization". Epigenetics & Chromatin. 4 (1): 13. doi:10.1186/1756-8935-4-13. PMC 3170176. PMID 21813016.
- Ravasi, Timothy; Suzuki, Harukazu; Cannistraci, Carlo Vittorio; et al. (March 2010). "An Atlas of Combinatorial Transcriptional Regulation in Mouse and Man". Cell. 140 (5): 744–752. doi:10.1016/j.cell.2010.01.044. PMC 2836267. PMID 20211142.
- Kratz, Anton; Arner, Erik; Saito, Rintaro; Kubosaki, Atsutaka; Kawai, Jun; Suzuki, Harukazu; Carninci, Piero; Arakawa, Takahiro; Tomita, Masaru; Hayashizaki, Yoshihide; Daub, Carsten O (2010). "Core promoter structure and genomic context reflect histone 3 lysine 9 acetylation patterns". BMC Genomics. 11 (1): 257. doi:10.1186/1471-2164-11-257. PMC 2867832. PMID 20409305.
- Akalin, Altuna; Fredman, David; Arner, Erik; Dong, Xianjun; Bryne, Jan; Suzuki, Harukazu; Daub, Carsten O; Hayashizaki, Yoshihide; Lenhard, Boris (2009). "Transcriptional features of genomic regulatory blocks". Genome Biology. 10 (4): R38. doi:10.1186/gb-2009-10-4-r38. PMC 2688929. PMID 19374772.
- "FANTOM 4 Publications". fantom.gsc.riken.jp.
- Noguchi, Shuhei; Arakawa, Takahiro; Fukuda, Shiro; et al. (29 August 2017). "FANTOM5 CAGE profiles of human and mouse samples". Scientific Data. 4: 170112. doi:10.1038/sdata.2017.112. PMC 5574368. PMID 28850106.
- Lizio, Marina; Harshbarger, Jayson; Shimoji, Hisashi; et al. (2015). "Gateways to the FANTOM5 promoter level mammalian expression atlas". Genome Biology. 16 (1): 22. doi:10.1186/s13059-014-0560-6. PMC 4310165. PMID 25723102.
- The FANTOM Consortium the RIKEN PMI CLST (DGT); Forrest, A. R.; Kawaji, H.; Rehli, M.; Baillie, J. K.; De Hoon, M. J.; Haberle, V.; Lassmann, T.; Kulakovskiy, I. V.; Lizio, M.; Itoh, M.; Andersson, R.; Mungall, C. J.; Meehan, T. F.; Schmeier, S.; Bertin, N.; Jørgensen, M.; Dimont, E.; Arner, E.; Schmidl, C.; Schaefer, U.; Medvedeva, Y. A.; Plessy, C.; Vitezic, M.; Severin, J.; Semple, C.; Ishizu, Y.; Young, R. S.; Francescatto, M.; et al. (27 March 2014). "A promoter-level mammalian expression atlas". Nature. 507 (7493): 462–470. doi:10.1038/nature13182. PMC 4529748. PMID 24670764.
- Andersson, Robin; Gebhard, Claudia; Miguel-Escalada, Irene; et al. (27 March 2014). "An atlas of active enhancers across human cell types and tissues". Nature. 507 (7493): 455–461. doi:10.1038/nature12787. PMC 5215096. PMID 24670763.
- Kanamori-Katayama, M.; Itoh, M.; Kawaji, H.; et al. (19 May 2011). "Unamplified cap analysis of gene expression on a single-molecule sequencer". Genome Research. 21 (7): 1150–1159. doi:10.1101/gr.115469.110. PMC 3129257. PMID 21596820.
- Arner, E.; Daub, C. O.; Vitting-Seerup, K.; et al. (12 February 2015). "Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells". Science. 347 (6225): 1010–1014. doi:10.1126/science.1259418. PMC 4681433. PMID 25678556.
- Severin, Jessica; Lizio, Marina; Harshbarger, Jayson; et al. (1 March 2014). "Interactive visualization and analysis of large-scale sequencing datasets using ZENBU". Nature Biotechnology. 32 (3): 217–219. doi:10.1038/nbt.2840. PMID 24727769.
- "FANTOM5_SSTAR". fantom.gsc.riken.jp.
- Ronnerblad, M.; Andersson, R.; Olofsson, T.; et al. (26 March 2014). "Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation". Blood. 123 (17): e79–e89. doi:10.1182/blood-2013-02-482893. PMID 24671952.
- Hon, Chung-Chau; Ramilowski, Jordan A.; Harshbarger, Jayson; et al. (1 March 2017). "An atlas of human long non-coding RNAs with accurate 5′ ends". Nature. 543 (7644): 199–204. doi:10.1038/nature21374. PMC 6857182. PMID 28241135.
- de Rie, Derek; Abugessaisa, Imad; Alam, Tanvir; et al. (21 August 2017). "An integrated expression atlas of miRNAs and their promoters in human and mouse". Nature Biotechnology. 35 (9): 872–878. doi:10.1038/nbt.3947. PMC 5767576. PMID 28829439.
- "FANTOM - FANTOM6". fantom.gsc.riken.jp.