Equilibrium constant
The equilibrium constant of a chemical reaction is the value of its reaction quotient at chemical equilibrium, a state approached by a dynamic chemical system after sufficient time has elapsed at which its composition has no measurable tendency towards further change. For a given set of reaction conditions, the equilibrium constant is independent of the initial analytical concentrations of the reactant and product species in the mixture. Thus, given the initial composition of a system, known equilibrium constant values can be used to determine the composition of the system at equilibrium. However, reaction parameters like temperature, solvent, and ionic strength may all influence the value of the equilibrium constant.
A knowledge of equilibrium constants is essential for the understanding of many chemical systems, as well as biochemical processes such as oxygen transport by hemoglobin in blood and acid-base homeostasis in the human body.
Stability constants, formation constants, binding constants, association constants and dissociation constants are all types of equilibrium constants.
Basic definitions and properties
For a system undergoing a reversible reaction described by the general chemical equation

a thermodynamic equilibrium constant, denoted by , is defined to be the value of the reaction quotient Qt when forward and reverse reactions occur at the same rate. At chemical equilibrium, the chemical composition of the mixture does not change with time and the Gibbs free energy change for the reaction is zero. If the composition of a mixture at equilibrium is changed by addition of some reagent, a new equilibrium position will be reached, given enough time. An equilibrium constant is related to the composition of the mixture at equilibrium by [1][2]




where {X} denotes the thermodynamic activity of reagent X at equilibrium, [X] the corresponding concentration, and γ the corresponding activity coefficient. If it can be assumed that the quotient of activity coefficients, , is constant over a range of experimental conditions, such as pH, then an equilibrium constant can be derived as a quotient of concentrations.


An equilibrium constant is related to the standard Gibbs free energy change of reaction by


where R is the universal gas constant, T is the absolute temperature (in kelvins), and ln is a natural logarithm. This expression implies that must be a pure number and cannot have a dimension, since logarithms can only be taken of pure numbers. must also be a pure number. On the other hand, the reaction quotient at equilibrium


does have the dimension of concentration raised to some power (see #Dimensionality, below). Such reaction quotients are often referred to, in the biochemical literature, as equilibrium constants.
For an equilibrium mixture of gases, an equilibrium constant can be defined in terms of partial pressure or fugacity.
An equilibrium constant is related to the forward and backward rate constants, kf and kr of the reactions involved in reaching equilibrium:

Types of equilibrium constants
Cumulative and stepwise formation constants
A cumulative or overall constant, given the symbol β, is the constant for the formation of a complex from reagents. For example, the cumulative constant for the formation of ML2 is given by
- M + 2 L ⇌ ML2; [ML2] = β12[M][L]2
The stepwise constant, K, for the formation of the same complex from ML and L is given by
- ML + L ⇌ ML2; [ML2] = K[ML][L] = Kβ11[M][L]2
It follows that
- β12 = Kβ11
A cumulative constant can always be expressed as the product of stepwise constants. There is no agreed notation for stepwise constants, though a symbol such as KL
ML is sometimes found in the literature. It is best always to define each stability constant by reference to an equilibrium expression.
Competition method
A particular use of a stepwise constant is in the determination of stability constant values outside the normal range for a given method. For example, EDTA complexes of many metals are outside the range for the potentiometric method. The stability constants for those complexes were determined by competition with a weaker ligand.
- ML + L′ ⇌ ML′ + L

The formation constant of [Pd(CN)4]2− was determined by the competition method.
Association and dissociation constants
In organic chemistry and biochemistry it is customary to use pKa values for acid dissociation equilibria.

where log denotes a logarithm to base 10 or common logarithm, and Kdiss is a stepwise acid dissociation constant. For bases, the base association constant, pKb is used. For any given acid or base the two constants are related by pKa + pKb = pKw, so pKa can always be used in calculations.
On the other hand, stability constants for metal complexes, and binding constants for host–guest complexes are generally expressed as association constants. When considering equilibria such as
- M + HL ⇌ ML + H
it is customary to use association constants for both ML and HL. Also, in generalized computer programs dealing with equilibrium constants it is general practice to use cumulative constants rather than stepwise constants and to omit ionic charges from equilibrium expressions. For example, if NTA, nitrilotriacetic acid, N(CH2CO2H)3 is designated as H3L and forms complexes ML and MHL with a metal ion M, the following expressions would apply for the dissociation constants.

The cumulative association constants can be expressed as

Note how the subscripts define the stoichiometry of the equilibrium product.
Micro-constants
When two or more sites in an asymmetrical molecule may be involved in an equilibrium reaction there are more than one possible equilibrium constants. For example, the molecule L-DOPA has two non-equivalent hydroxyl groups which may be deprotonated. Denoting L-DOPA as LH2, the following diagram shows all the species that may be formed (X = CH2CH(NH2)CO2H):
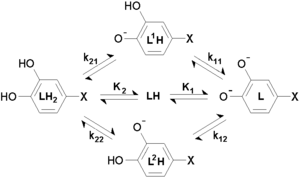
The concentration of the species LH is equal to the sum of the concentrations of the two micro-species with the same chemical formula, labelled L1H and L2H. The constant K2 is for a reaction with these two micro-species as products, so that [LH] = [L1H] + [L2H] appears in the numerator, and it follows that this macro-constant is equal to the sum of the two micro-constants for the component reactions.
- K2 = k21 + k22
However, the constant K1 is for a reaction with these two micro-species as reactants, and [LH] = [L1H] + [L2H] in the denominator, so that in this case[3]
- 1/K1 =1/ k11 + 1/k12,
and therefore K1 =k11 k12 / (k11 + k12). Thus, in this example there are four micro-constants whose values are subject to two constraints; in consequence, only the two macro-constant values, for K1 and K2 can be derived from experimental data.
Micro-constant values can, in principle, be determined using a spectroscopic technique, such as infrared spectroscopy, where each micro-species gives a different signal. Methods which have been used to estimate micro-constant values include
- Chemical: blocking one of the sites, for example by methylation of a hydroxyl group, followed by determination of the equilibrium constant of the related molecule, from which the micro-constant value for the "parent" molecule may be estimated.
- Mathematical: applying numerical procedures procedures to 13C NMR data.[4][5]
Although the value of a micro-constant cannot be determined from experimental data, site occupancy, which is proportional to the micro-constant value can be very important for biological activity. Therefore various methods have been developed for estimating micro-constant values. For example, the isomerization constant for L-DOPA has been estimated to have a value of 0.9, so the micro-species L1H and L2H have almost equal concentrations at all pH values.
pH considerations (Brønsted constants)
pH is defined in terms of the activity of the hydrogen ion
- pH = −log10 {H+}
In the approximation of ideal behaviour, activity is replaced by concentration. pH is measured by means of a glass electrode, a mixed equilibrium constant, also known as a Brønsted constant, may result.
- HL ⇌ L + H;

It all depends on whether the electrode is calibrated by reference to solutions of known activity or known concentration. In the latter case the equilibrium constant would be a concentration quotient. If the electrode is calibrated in terms of known hydrogen ion concentrations it would be better to write p[H] rather than pH, but this suggestion is not generally adopted.
Hydrolysis constants
In aqueous solution the concentration of the hydroxide ion is related to the concentration of the hydrogen ion by


The first step in metal ion hydrolysis[6] can be expressed in two different ways

It follows that β* = KKW. Hydrolysis constants are usually reported in the β* form and therefore often have values much less than 1. For example, if log K = 4 and log KW = −14, log β* = 4 + (−14) = −10 so that β* = 10−10. In general when the hydrolysis product contains n hydroxide groups log β* = log K + n log KW
Conditional constants
Conditional constants, also known as apparent constants, are concentration quotients which are not true equilibrium constants but can be derived from them.[7] A very common instance is where pH is fixed at a particular value. For example, in the case of iron(III) interacting with EDTA, a conditional constant could be defined by

This conditional constant will vary with pH. It has a maximum at a certain pH. That is the pH where the ligand sequesters the metal most effectively.
In biochemistry equilibrium constants are often measured at a pH fixed by means of a buffer solution. Such constants are, by definition, conditional and different values may be obtained when using different buffers.
Gas-phase equilibria
For equilibria in a gas phase, fugacity, f, is used in place of activity. However, fugacity has the dimension of pressure, so it must be divided by a standard pressure, usually 1 bar, in order to produce a dimensionless quantity, f/po. An equilibrium constant is expressed in terms of the dimensionless quantity. For example, for the equilibrium 2NO2 ⇌ N2O4,

Fugacity is related to partial pressure, p, by a dimensionless fugacity coefficient ϕ: f = ϕp. Thus, for the example,

Usually the standard pressure is omitted from such expressions. Expressions for equilibrium constants in the gas phase then resemble the expression for solution equilibria with fugacity coefficient in place of activity coefficient and partial pressure in place of concentration.

Thermodynamic basis for equilibrium constant expressions
Thermodynamic equilibrium is characterized by the free energy for the whole (closed) system being a minimum. For systems at constant temperature and pressure the Gibbs free energy is minimum.[8] The slope of the reaction free energy with respect to the reaction coordinate, ξ, is zero when the free energy is at its minimum value.

The free energy change, δGr, can be expressed as a weighted sum of chemical potentials, the partial molar free energies of the species in equilibrium. The potential, μi, of the ith species in a chemical reaction is the partial derivative of the free energy with respect to the number of moles of that species, Ni

A general chemical equilibrium can be written as

where nj are the stoichiometric coefficients of the reactants in the equilibrium equation, and mj are the coefficients of the products. At equilibrium

The chemical potential, μi, of the ith species can be calculated in terms of its activity, ai.

μo
i is the standard chemical potential of the species, R is the gas constant and T is the temperature. Setting the sum for the reactants j to be equal to the sum for the products, k, so that δGr(Eq) = 0

Rearranging the terms,


This relates the standard Gibbs free energy change, ΔGo to an equilibrium constant, K, the reaction quotient of activity values at equilibrium.


Equivalence of thermodynamic and kinetic expressions for equilibrium constants
At equilibrium the rate of the forward reaction is equal to the backward reaction rate. A simple reaction, such as ester hydrolysis

has reaction rates given by expressions


According to Guldberg and Waage, equilibrium is attained when the forward and backward reaction rates are equal to each other. In these circumstances, an equilibrium constant is defined to be equal to the ratio of the forward and backward reaction rate constants.

For convenience the concentration of water may be taken to be constant, resulting in the simpler expression

This particular concentration quotient, , has the dimension of concentration, but the thermodynamic equilibrium constant, K, is always dimensionless.

Unknown activity coefficient values
It is very rare for activity coefficient values to have been determined experimentally for a system at equilibrium. There are three options for dealing with the situation where activity coefficient values are not known from experimental measurements.
- Use calculated activity coefficients, together with concentrations of reactants. For equilibria in solution estimates of the activity coefficients of charged species can be obtained using Debye–Hückel theory, an extended version, or SIT theory. For uncharged species, the activity coefficient γ0 mostly follows a "salting-out" model: log10 γ0 = bI where I stands for ionic strength.[9]
- Assume that the activity coefficients are all equal to 1. This is acceptable when all concentrations are very low.
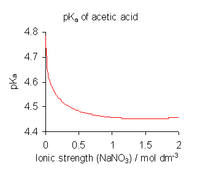
- For equilibria in solution use a medium of high ionic strength. In effect this redefines the standard state as referring to the medium. Activity coefficients in the standard state are, by definition, equal to 1. The value of an equilibrium constant determined in this manner is dependent on the ionic strength. When published constants refer to an ionic strength other than the one required for a particular application, they may be adjusted by means of specific ion theory (SIT) and other theories.[10]
Dimensionality
An equilibrium constant is related to the standard Gibbs free energy change, , for the reaction by the expression

Therefore, K, must be a number from which a logarithm can be derived. In the case of a simple equilibrium

the thermodynamic equilibrium constant is defined in terms of the activities, {AB}, {A} and {B}, of the species in equilibrium with each other.

Now, each activity term can be expressed as a product of a concentration and a corresponding activity coefficient, . Therefore,



When the quotient of activity coefficients, is set equal to 1

K then appears to have the dimension of 1/concentration. This is what usually happens in practice when an equilibrium constant is calculated as a quotient of concentration values. The assumption underlying this practice is that the quotient of activities is constant under the conditions in which the equilibrium constant value is determined. These conditions are usually achieved by keeping the reaction temperature constant and by using a medium of relatively high ionic strength as the solvent. It is not unusual, particularly in texts relating to biochemical equilibria, to see a value quoted with a dimension. The justification for this practice is that the concentration scale used may be either mol dm−3 or mmol dm−3, so that the concentration unit has to be stated in order to avoid there being any ambiguity.
Note. When the concentration values are measured on the mole fraction scale all concentrations and activity coefficients are dimensionless quantities.
In general equilibria between two reagents can be expressed as

in which case the equilibrium constant is defined, in terms of numerical concentration values, as

The apparent dimension of this K value is concentration1−p−q; this may be written as M(1−p−q) or mM(1−p−q), where the symbol M signifies a molar concentration (1M = 1 mol dm−3). The apparent dimension of a dissociation constant is the reciprocal of the apparent dimension of the corresponding association constant, and vice versa.
When discussing the thermodynamics of chemical equilibria it is necessary to take dimensionality into account. There are two possible approaches.
- Set the dimension of Γ to be the reciprocal of the dimension of the concentration quotient. This is almost universal practice in the field of stability constant determinations. The "equilibrium constant" , is dimensionless. It will be a function of the ionic strength of the medium used for the determination. Setting the numerical value of Γ to be 1 is equivalent to re-defining the standard states.
- Replace each concentration term by the dimensionless quotient , where is the concentration of reagent X in its standard state. By definition the numerical value of is 1, so Γ also has a numerical value of 1.




In both approaches the numerical value of the stability constant is unchanged. The first is more useful for practical purposes; in fact, the unit of the concentration quotient is often attached to a published stability constant value in the biochemical literature. The second approach is consistent with the standard exposition of Debye–Hückel theory, where , etc. are taken to be pure numbers.

Water as both reactant and solvent
For reactions in aqueous solution, such as an acid dissociation reaction
- AH + H2O ⇌ A− + H3O+
the concentration of water may be taken as being constant and the formation of the hydronium ion is implicit.
- AH ⇌ A− + H+
Water concentration is omitted from expressions defining equilibrium constants, except when solutions are very concentrated.
- (K defined as a dissociation constant)

Similar considerations apply to metal ion hydrolysis reactions.
Enthalpy and entropy: temperature dependence
If both the equilibrium constant, and the standard enthalpy change, , for a reaction have been determined experimentally, the standard entropy change for the reaction is easily derived. Since and





To a first approximation the standard enthalpy change is independent of temperature. Using this approximation, definite integration of the van 't Hoff equation

gives[11]

This equation can be used to calculate the value of log K at a temperature, T2, knowing the value at temperature T1.
The van 't Hoff equation also shows that, for an exothermic reaction (), when temperature increases K decreases and when temperature decreases K increases, in accordance with Le Chatelier's principle. The reverse applies when the reaction is endothermic.

When K has been determined at more than two temperatures, a straight line fitting procedure may be applied to a plot of against to obtain a value for . Error propagation theory can be used to show that, with this procedure, the error on the calculated value is much greater than the error on individual log K values. Consequently, K needs to be determined to high precision when using this method.For example, with a silver ion-selective electrode each log K value was determined with a precision of ca. 0.001 and the method was applied successfully.[12]


Standard thermodynamic arguments can be used to show that, more generally, enthalpy will change with temperature.

where Cp is the heat capacity at constant pressure.
A more complex formulation
The calculation of K at a particular temperature from a known K at another given temperature can be approached as follows if standard thermodynamic properties are available. The effect of temperature on equilibrium constant is equivalent to the effect of temperature on Gibbs energy because:

where ΔrGo is the reaction standard Gibbs energy, which is the sum of the standard Gibbs energies of the reaction products minus the sum of standard Gibbs energies of reactants.
Here, the term "standard" denotes the ideal behaviour (i.e., an infinite dilution) and a hypothetical standard concentration (typically 1 mol/kg). It does not imply any particular temperature or pressure because, although contrary to IUPAC recommendation, it is more convenient when describing aqueous systems over wide temperature and pressure ranges.[13]
The standard Gibbs energy (for each species or for the entire reaction) can be represented (from the basic definitions) as:

In the above equation, the effect of temperature on Gibbs energy (and thus on the equilibrium constant) is ascribed entirely to heat capacity. To evaluate the integrals in this equation, the form of the dependence of heat capacity on temperature needs to be known.
If the standard molar heat capacity Co
p can be approximated by some analytic function of temperature (e.g. the Shomate equation), then the integrals involved in calculating other parameters may be solved to yield analytic expressions for them. For example, using approximations of the following forms:[14]
- For pure substances (solids, gas, liquid):

- For ionic species at T < 200 °C:

then the integrals can be evaluated and the following final form is obtained:

The constants A, B, C, a, b and the absolute entropy, S̆ o
298 K, required for evaluation of Co
p(T), as well as the values of G298 K and S298 K for many species are tabulated in the literature.
Pressure dependence
The pressure dependence of the equilibrium constant is usually weak in the range of pressures normally encountered in industry, and therefore, it is usually neglected in practice. This is true for condensed reactant/products (i.e., when reactants and products are solids or liquid) as well as gaseous ones.
For a gaseous-reaction example, one may consider the well-studied reaction of hydrogen with nitrogen to produce ammonia:
- N2 + 3 H2 ⇌ 2 NH3
If the pressure is increased by the addition of an inert gas, then neither the composition at equilibrium nor the equilibrium constant are appreciably affected (because the partial pressures remain constant, assuming an ideal-gas behaviour of all gases involved). However, the composition at equilibrium will depend appreciably on pressure when:
- the pressure is changed by compression or expansion of the gaseous reacting system, and
- the reaction results in the change of the number of moles of gas in the system.
In the example reaction above, the number of moles changes from 4 to 2, and an increase of pressure by system compression will result in appreciably more ammonia in the equilibrium mixture. In the general case of a gaseous reaction:
- α A + β B ⇌ σ S + τ T
the change of mixture composition with pressure can be quantified using:

where p denote the partial pressures and X the mole fractions of the components, P is the total system pressure, Kp is the equilibrium constant expressed in terms of partial pressures and KX is the equilibrium constant expressed in terms of mole fractions.
The above change in composition is in accordance with Le Chatelier's principle and does not involve any change of the equilibrium constant with the total system pressure. Indeed, for ideal-gas reactions Kp is independent of pressure.[15]

In a condensed phase, the pressure dependence of the equilibrium constant is associated with the reaction volume.[16] For reaction:
- α A + β B ⇌ σ S + τ T
the reaction volume is:

where V̄ denotes a partial molar volume of a reactant or a product.
For the above reaction, one can expect the change of the reaction equilibrium constant (based either on mole-fraction or molal-concentration scale) with pressure at constant temperature to be:

The matter is complicated as partial molar volume is itself dependent on pressure.
Effect of isotopic substitution
Isotopic substitution can lead to changes in the values of equilibrium constants, especially if hydrogen is replaced by deuterium (or tritium).[17] This equilibrium isotope effect is analogous to the kinetic isotope effect on rate constants, and is primarily due to the change in zero-point vibrational energy of H–X bonds due to the change in mass upon isotopic substitution.[17] The zero-point energy is inversely proportional to the square root of the mass of the vibrating hydrogen atom, and will therefore be smaller for a D–X bond that for an H–X bond.
An example is a hydrogen atom abstraction reaction R' + H–R ⇌ R'–H + R with equilibrium constant KH, where R' and R are organic radicals such that R' forms a stronger bond to hydrogen than does R. The decrease in zero-point energy due to deuterium substitution will then be more important for R'–H than for R–H, and R'–D will be stabilized more than R–D, so that the equilibrium constant KD for R' + D–R ⇌ R'–D + R is greater than KH. This is summarized in the rule the heavier atom favors the stronger bond.[17]
Similar effects occur in solution for acid dissociation constants (Ka) which describe the transfer of H+ or D+ from a weak aqueous acid to a solvent molecule: HA + H2O = H3O+ + A− or DA + D2O ⇌ D3O+ + A−. The deuterated acid is studied in heavy water, since if it were dissolved in ordinary water the deuterium would rapidly exchange with hydrogen in the solvent.[17]
The product species H3O+ (or D3O+) is a stronger acid than the solute acid, so that it dissociates more easily and its H–O (or D–O) bond is weaker than the H–A (or D–A) bond of the solute acid. The decrease in zero-point energy due to isotopic substitution is therefore less important in D3O+ than in DA so that KD < KH, and the deuterated acid in D2O is weaker than the non-deuterated acid in H2O. In many cases the difference of logarithmic constants pKD – pKH is about 0.6,[17] so that the pD corresponding to 50% dissociation of the deuterated acid is about 0.6 units higher than the pH for 50% disssociation of the non-deuterated acid.
See also
References
- IUPAC Gold Book.
- Rossotti, F. J. C.; Rossotti, H. (1961). The Determination of Stability Constants. McGraw-Hill. p. 5.
- Splittgerber, A. G.; Chinander, L.L. (1 February 1988). "The spectrum of a dissociation intermediate of cysteine: a biophysical chemistry experiment". Journal of Chemical Education. 65 (2): 167.
- Hague, David N.; Moreton, Anthony D. (1994). "Protonation sequence of linear aliphatic polyamines by 13C NMR spectroscopy". J. Chem. Soc., Perkin Trans. 2 (2): 265–70. doi:10.1039/P29940000265.
- Borkovec, Michal; Koper, Ger J. M. (2000). "A Cluster Expansion Method for the Complete Resolution of Microscopic Ionization Equilibria from NMR Titrations". Anal. Chem. 72 (14): 3272–9. doi:10.1021/ac991494p. PMID 10939399.
- Baes, C. F.; Mesmer, R. E. (1976). The Hydrolysis of Cations. Wiley.
- Schwarzenbach, G.; Flaschka, H. (1969). Complexometric titrations. Methuen.
- Denbigh, K. (1981). "Chapter 4". The principles of chemical equilibrium (4th ed.). Cambridge: Cambridge University Press. ISBN 978-0-521-28150-8.
- Butler, J. N. (1998). Ionic Equilibrium. John Wiley and Sons.
- "Project: Ionic Strength Corrections for Stability Constants". International Union of Pure and Applied Chemistry. Archived from the original on 29 October 2008. Retrieved 2008-11-23.
- Atkins, Peter; de Paula, Julio (2006). Physical Chemistry. Oxford. p. 214. ISBN 978-0198700722.
- Barnes, D.S.; Ford, G.J; Pettit, L.D.; Sherringham, C. (1971). "Ligands containing elements of group VIB. Part V. Thermodynamics of silver complex formation of some saturated and unsaturated (alkyl-thio)acetic and (alkylseleno)acetic acids". J. Chem. Soc. A: 2883–2887. doi:10.1039/J19710002883.
- Majer, V.; Sedelbauer, J.; Wood (2004). "Calculations of standard thermodynamic properties of aqueous electrolytes and nonelectrolytes". In Palmer, D. A.; Fernandez-Prini, R.; Harvey, A. (eds.). Aqueous Systems at Elevated Temperatures and Pressures: Physical Chemistry of Water, Steam and Hydrothermal Solutions. Elsevier.
- Roberge, P. R. (November 2011). "Appendix F". Handbook of Corrosion Engineering. McGraw-Hill. p. 1037ff.
- Atkins, P. W. (1978). Physical Chemistry (6th ed.). Oxford University Press. p. 210.
- Van Eldik, R.; Asano, T.; Le Noble, W. J. (1989). "Activation and reaction volumes in solution. 2". Chem. Rev. 89 (3): 549–688. doi:10.1021/cr00093a005.
- Laidler K.J. Chemical Kinetics (3rd ed., Harper & Row 1987), p.428–433 ISBN 0-06-043862-2
Data sources
- IUPAC SC-Database A comprehensive database of published data on equilibrium constants of metal complexes and ligands
- NIST Standard Reference Database 46: Critically selected stability constants of metal complexes
- Inorganic and organic acids and bases pKa data in water and DMSO
- NASA Glenn Thermodynamic Database webpage with links to (self-consistent) temperature-dependent specific heat, enthalpy, and entropy for elements and molecules