Epidermolysis bullosa
Epidermolysis bullosa (EB) is a group of rare genetic conditions that result in easy blistering of the skin and mucous membranes.[2][3] Blisters occur with minor trauma or friction and are painful.[2] Its severity can range from mild to fatal.[2] Those with mild cases may not develop symptoms until they start to crawl or walk.[7] Complications may include esophageal narrowing, squamous cell skin cancer, and the need for amputations.[4][5]
| Epidermolysis bullosa | |
|---|---|
| Other names | Butterfly children[1] |
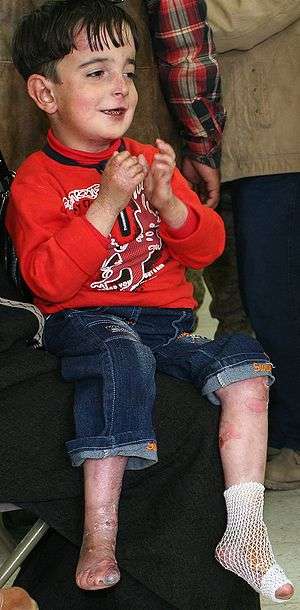 | |
| A five-year-old boy with epidermolysis bullosa | |
| Specialty | Dermatology |
| Symptoms | Painful skin blisters[2][3] |
| Complications | Esophageal narrowing, squamous cell skin cancer, amputations[4][5] |
| Usual onset | At birth[5] |
| Duration | Often lifelong[5] |
| Types | Epidermolysis bullosa simplex, dystrophic epidermolysis bullosa, junctional epidermolysis bullosa, Kindler syndrome[2] |
| Causes | Genetic[2] |
| Diagnostic method | Skin biopsy, genetic testing[6] |
| Differential diagnosis | Bullous pemphigoid, pemphigus vulgaris, friction blisters, insect bites[5] |
| Treatment | Wound care, pain control, controlling infections, nutritional support[2] |
| Frequency | c. 500,000[5] |
EB is due to a mutation in at least one of 18 different genes.[2] Some types are autosomal dominant while others are autosomal recessive.[2] The underlying mechanism is a defect in attachment between or within the layers of the skin.[5] There are four main types: epidermolysis bullosa simplex, dystrophic epidermolysis bullosa, junctional epidermolysis bullosa, and Kindler syndrome.[2] The diagnosis is suspected based on symptoms and confirmed by skin biopsy or genetic testing.[6][5]
There is no cure for the condition.[4] Management involves wound care, pain control, controlling infections, nutritional support, and prevention and treatment of complications.[2] About half a million people are affected globally.[5] It occurs equally commonly in males and females.[7] Children who are affected may be bullied by other children or experience inappropriate comments from adults.[5]
Classification
Epidermolysis bullosa refers to a group of disorders that involve the formation of blisters following trivial trauma. Over 300 mutations have been identified in this condition.[8] They have been classified into the following types:[9][10]:596
Epidermolysis bullosa simplex
Epidermolysis bullosa simplex is a form of epidermolysis bullosa that causes blisters at the site of rubbing. It typically affects the hands and feet, and is typically inherited in an autosomal dominant manner, affecting the keratin genes KRT5 and KRT14. Therefore, there is a failure in keratinisation, which affects the integrity and the ability of the skin to resist mechanical stresses.
Junctional epidermolysis bullosa
Junctional epidermolysis bullosa is an inherited disease affecting laminin and collagen. This disease is characterised by blister formation within the lamina lucida of the basement membrane zone[10]:599 and is inherited in an autosomal recessive manner. It also presents with blisters at the site of friction, especially on the hands and feet, and has variants that can occur in children and adults. Less than one person per million people is estimated to have this form of epidemolysis bullosa.[11]
Dystrophic epidermolysis bullosa
Dystrophic epidermolysis bullosa is an inherited variant affecting the skin and other organs. Dystrophic epidermolysis bullosa is caused by genetic defects (or mutations) within the human COL7A1 gene encoding the protein type VII collagen (collagen VII).[12] DEB-causing mutations can be either autosomal dominant or autosomal recessive. Epidermis bullosa pruriginosa and Albopapuloid epidermolysis bullosa (Pasini's disease) are rare subtypes of this disease.[13]
Epidermolysis bullosa acquisita
Acral peeling
Pathophysiology
The human skin consists of two layers: an outermost layer called the epidermis and a layer underneath called the dermis. In individuals with healthy skin, there are protein anchors between these two layers that prevent them from moving independently from one another (shearing). In people born with EB, the two skin layers lack the protein anchors that hold them together, resulting in extremely fragile skin—even minor mechanical friction (like rubbing or pressure) or trauma will separate the layers of the skin and form blisters and painful sores. Sufferers of EB have compared the sores with third-degree burns. Furthermore, as a complication of the chronic skin damage, people suffering from EB have an increased risk of malignancies (cancers) of the skin.
Diagnosis
Epidermolysis bullosa can be diagnosed either by a skin (punch) biopsy at the edge of a wound with immunofluorescent mapping, or via blood sample and genetic testing.
Treatment
Recent research has focused on changing the mixture of keratins produced in the skin. There are 54 known keratin genes—of which 28 belong to the type I intermediate filament genes and 26 to type II—which work as heterodimers. Many of these genes share substantial structural and functional similarity, but they are specialized to cell type and/or conditions under which they are normally produced. If the balance of production could be shifted away from the mutated, dysfunctional keratin gene toward an intact keratin gene, symptoms could be reduced. For example, sulforaphane, a compound found in broccoli, was found to reduce blistering in a mouse model to the point where affected pups could not be identified visually, when injected into pregnant mice (5 µmol/day = 0.9 mg) and applied topically to newborns (1 µmol/day = 0.2 mg in jojoba oil).[14]
As of 2008 clinical research at the University of Minnesota has explored allogeneic bone marrow transplantation for RD and junctional EB, treating a 2-year-old child who is one of 2 brothers with EB. A second transplant has also been performed on the child's older brother, and a third transplant is scheduled for a California baby. A clinical trial is planned for 30 subjects.[15] However, the immune suppression that bone marrow transplantation requires causes a risk of serious infections with large scale blisters and skin erosion.[16] Indeed, at least four people have died in the course of either preparation for or institution of bone marrow transplantation for epidermolysis bullosa, out of only a small group of patients treated so far.[16] The mechanism of action of this therapy is unclear as hematopoietic stem cells are not thought to contribute to epithelial lineages. Rather, it is speculated that cross-correction from tissue-resident graft-derived immune cells contributes to the observed clinical benefit.[17]
A pilot study performed in 2015 suggests that systemic granulocyte-colony stimulating factor (G-CSF) may promote increased wound healing in people with dystrophic epidermolysis bullosa.[18] Transplanting skin derived from genetically modified stem cells onto the wound surfaces has been studied with a report of improvements in one person.[19]
Monitoring
The Epidermolysis Bullosa Disease Activity and Scarring index (EBDASI) is a scoring system that objectively quantifies the severity of epidermolysis bullosa. The EBDASI is a tool for clinicians and patients to monitor the severity of the disease. It has also been designed to evaluate the response to new therapies for the treatment of EB. The EBDASI was developed and validated by Professor Dedee Murrell and her team of students and fellows at the St George Hospital, University of New South Wales, in Sydney, Australia. It was presented at the International Investigative Dermatology congress in Edinburgh in 2013 and a paper-based version was published in the Journal of the American Academy of Dermatology in 2014.[20]
Prognosis
A 2014 study classified cases into three types—epidermolysis bullosa simplex (EBS), junctional epidermolysis bullosa (JEB), and dystrophic epidermolysis bullosa (DEB) -- and reviewed their times of death. The first two types tended to die in infancy and the last in early adulthood.[21] In a survey of 11 families affected by the disease, lack of awareness of the disease by both the public and health care providers raised concerns about the care provided as well as insensitive comments by adults and bullying by children.[5]
Epidemiology
An estimated 20 per million live births are diagnosed with EB,[22] and 9 per million people in the general population have the condition. Of these cases, approximately 92% are epidermolysis bullosa simplex (EBS), 5% are dystrophic epidermolysis bullosa (DEB), 1% are junctional epidermolysis bullosa (JEB), and 2% are unclassified. Carrier frequency ranges from 1 in 333 for JEB, to 1 in 450 for DEB; the carrier frequency for EBS is presumed to be much higher than JEB or DEB.
The disorder occurs in every racial and ethnic group and affects both sexes.[23][24]
Society and culture
In 2010, Emma Fogarty, a campaigner for DEBRA Ireland (the EB charity) was awarded a People of the Year Award.[25] Actor Colin Farrell has campaigned with Fogarty on behalf of sufferers.[26]
In 2014, Pearl Jam lead vocalist Eddie Vedder together with his wife Jill McCormick co-founded the EB Research Partnership,[27] a non-profit organization dedicated to finding a cure for epidermolysis bullosa.[28] McCormick is childhood friends with Ryan Fullmer, whose son, Michael, was born with EB. Vedder, McCormick, Ryan Fullmer, and his wife, Heather founded Heal EB. In 2014, they merged Heal EB with the Jackson Gabriel Research Foundation to create the EB Research Partnership. The EBRP hosts several annual fundraising events. To date, they have raised $12 million to fund research to find a cure.[29] On March 1, 2019, heavyweight boxer Luis Ortiz was named an honorary ambassador for the EB community by the EB Research Partnership. Ortiz's daughter, Lismercedes, was born with epidermolysis bullosa.[30]
Movies
The condition was brought to public attention in 2004 in the UK through the Channel 4 documentary The Boy Whose Skin Fell Off, chronicling the life and death of Jonny Kennedy, an Englishman with EB.[31] In the United States, the same could be said of the HBO documentary My Flesh and Blood from 2003. Additionally the film, Butterfly Girl, follows Abigail Evans with the disease.[32] In Canada, The Sports Network's award-winning documentary on Jonathan Pitre led to extensive coverage on the boy's disease, treatment, and death.[33][34]
Other names
Other terms used to describe those infected include "butterfly children" as the skin is fragile as a butterfly’s wings,[35] "cotton wool babies",[36][37] or "crystal skin children".[38]
References
- Fine, Jo-David; Hintner, Helmut (2009). Life with Epidermolysis Bullosa (EB): Etiology, Diagnosis, Multidisciplinary Care and Therapy. Springer Science & Business Media. p. 242. ISBN 9783211792711.
- "Epidermolysis bullosa". rarediseases.info.nih.gov. Retrieved 16 May 2018.
- "Epidermolysis Bullosa". National Institute of Arthritis and Musculoskeletal and Skin Diseases. 11 April 2017. Retrieved 16 May 2018.
- "Epidermolysis Bullosa". National Institute of Arthritis and Musculoskeletal and Skin Diseases. 11 April 2017. Retrieved 16 May 2018.
- Tabor, A; Pergolizzi JV, Jr; Marti, G; Harmon, J; Cohen, B; Lequang, JA (May 2017). "Raising Awareness Among Healthcare Providers about Epidermolysis Bullosa and Advancing Toward a Cure". The Journal of Clinical and Aesthetic Dermatology. 10 (5): 36–48. PMC 5479476. PMID 28670357.
- "Epidermolysis Bullosa". National Institute of Arthritis and Musculoskeletal and Skin Diseases. 11 April 2017. Retrieved 16 May 2018.
- "Epidermolysis Bullosa". National Institute of Arthritis and Musculoskeletal and Skin Diseases. 11 April 2017. Retrieved 16 May 2018.
- Koshida, S.; Tsukamura, A.; Yanagi, T.; Nakahara, S.; Takeuchi, Y.; Kato, T.; Tanaka, T.; Nakano, H.; Shimizu, H. (2013). "Hallopeau-Siemens dystrophic epidermolysis bullosa due to homozygous 5818delC mutation in the COL7A gene". Pediatr Int. 55 (2): 234–7. doi:10.1111/j.1442-200X.2012.03638.x. PMID 23679163.
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0-07-138076-0.
- "Junctional epidermolysis bullosa". Genetics Home Reference. NIH. Retrieved 11 October 2013.
- Varki, Roslyn; Sadowski, Sara; Uitto, Jouni; Pfendner, Ellen (2007-03-01). "Epidermolysis bullosa. II. Type VII collagen mutations and phenotype–genotype correlations in the dystrophic subtypes". Journal of Medical Genetics. 44 (3): 181–192. doi:10.1136/jmg.2006.045302. ISSN 0022-2593. PMC 2598021. PMID 16971478.
- McGRATH, J.a.; Schofield, O.m.v.; Eady, R.a.j. (1994-05-01). "Epidermolysis bullosa pruriginosa: dystrophic epidermolysis bullosa with distinctive clinicopathological features". British Journal of Dermatology. 130 (5): 617–625. doi:10.1111/j.1365-2133.1994.tb13109.x. ISSN 1365-2133. PMID 8204470.
- Kerns, Michelle L.; DePianto, Daryle; Dinkova-Kostova, Albena T.; Talalay, Paul; Coulombe, Pierre A. (2007-09-04). "Reprogramming of keratin biosynthesis by sulforaphane restores skin integrity in epidermolysis bullosa simplex". Proceedings of the National Academy of Sciences. 104 (36): 14460–14465. Bibcode:2007PNAS..10414460K. doi:10.1073/pnas.0706486104. ISSN 0027-8424. PMC 1964870. PMID 17724334.
- Josephine Marcotty (2008-07-03). "Long-shot stem-cell treatment gives two brothers a future". Star Tribune. Retrieved 2008-07-22.
- Souillet, G.; Guffon, N.; Maire, I.; Pujol, M.; Taylor, P.; Sevin, F.; Bleyzac, N.; Mulier, C.; Durin, A. (2003-06-10). "Outcome of 27 patients with Hurler's syndrome transplanted from either related or unrelated haematopoietic stem cell sources". Bone Marrow Transplantation. 31 (12): 1105–17. doi:10.1038/sj.bmt.1704105. PMID 12796790.
- Wagner, John E.; Ishida-Yamamoto, Akemi; McGrath, John A.; Hordinsky, Maria; Keene, Douglas R.; Riddle, Megan J.; Osborn, Mark J.; Lund, Troy; Dolan, Michelle; Blazar, Bruce R.; Tolar, Jakub (2010-08-12). "Bone Marrow Transplantation for Recessive Dystrophic Epidermolysis Bullosa". The New England Journal of Medicine. 363 (7): 629–639. doi:10.1056/NEJMoa0910501. ISSN 0028-4793. PMC 2967187. PMID 20818854.
- Fine, Jo-David; Manes, Becky; Frangoul, Haydar (July 2015). "Systemic granulocyte colony-stimulating factor (G-CSF) enhances wound healing in dystrophic epidermolysis bullosa (DEB): Results of a pilot trial". Journal of the American Academy of Dermatology. 73 (1): 56–61. doi:10.1016/j.jaad.2015.04.015. ISSN 0190-9622. PMID 25956659.
- Hirsch, T; Rothoeft, T; Teig, N; Bauer, JW; Pellegrini, G; De Rosa, L; Scaglione, D; Reichelt, J; Klausegger, A; Kneisz, D; Romano, O; Secone Seconetti, A; Contin, R; Enzo, E; Jurman, I; Carulli, S; Jacobsen, F; Luecke, T; Lehnhardt, M; Fischer, M; Kueckelhaus, M; Quaglino, D; Morgante, M; Bicciato, S; Bondanza, S; De Luca, M (16 November 2017). "Regeneration of the entire human epidermis using transgenic stem cells". Nature. 551 (7680): 327–332. Bibcode:2017Natur.551..327H. doi:10.1038/nature24487. PMC 6283270. PMID 29144448.
- Development, reliability, and validity of a novel Epidermolysis Bullosa Disease Activity and Scarring index (EBDASI). 2014. Loh CH, Kim J, Su JC, Daniel BS, Venugopal SS, Rhodes LM, Intong LR, Law MG, Murrell DF. January 2014. JAAD, 70, 1pp.
- Hon, Kam Lun Ellis; Li, Joshua J.; Cheng, Bernadette L.; Luk, David C.; Murrell, Dedee F.; Choi, Paul C. L.; Leung, Alexander K. C. (2015-03-04). "Age and etiology of childhood epidermolysis bullosa mortality". Journal of Dermatological Treatment. 26 (2): 178–182. doi:10.3109/09546634.2014.915002. ISSN 0954-6634. PMID 24724596.
- "Epidermolysis Bullosa Clinic Frequently Asked Questions". Stanford Medicine — Dermatology. Retrieved 13 April 2018.
- Marinkovich, MP; Wells, MJ; Perry, V; James, WD; Ortonne, JP (December 3, 2007). "Epidermolysis Bullosa". Bullous Diseases. emedicine.com. Retrieved 2008-07-22.
- Pfendner, Ellen; Uitto, Jouni; Fine, Jo-David (2000-11-07). "Epidermolysis Bullosa Carrier Frequencies in the US Population". Journal of Investigative Dermatology. 116 (3): 483–484. doi:10.1046/j.1523-1747.2001.127911.x. Retrieved 2008-07-22.
- Kiernan, Lynda (21 December 2018). "A brave Laois woman is among the guests for tonight's Late Late Show". Leinster Express. Retrieved 21 December 2018.
- Townsend, Michelle (21 December 2018). "Late Late Show viewers praise Colin Farrell and EB sufferer Emma Fogarty for honest interview". RSVP live. Archived from the original on 22 December 2018.
- "Board of Directors - EB Research Partnership". ebresearch.org.
- Reed, Ryan (2014-10-10). "Eddie Vedder Supports Research for Curing Rare Skin Disease EB". Rolling Stone.
- Lee, Kate (November 11, 2016). "Eddie Vedder and the Chicago Cubs Team Up to Raise $50,000 to Help Kids with EB". Medium.
- "Luis Ortiz named ambassador for Epidermolysis Bullosa Research Partnership". World Boxing News. March 1, 2009.
- "Series 1 - The Boy Whose Skin Fell Off". Channel 4. 2004. Retrieved 2009-02-28.
- Butterfly Girl: Remembering Abigail Evans, The Austinot, January 12, 2015.
- Andrew Duffy, "TSN documentary on Jonathan Pitre wins screen award". Ottawa Citizen. March 9, 2016. Retrieved 6 April 2018.
- Ottawa boy Jonathan Pitre dead at 17 from rare skin disease, CBC News, April 6, 2018.
- Roddy Isles; Head of Press (2005-05-12). "Dundee Scientists on road to cure for "Butterfly Children" condition". University of Dundee. Retrieved 2008-07-22.
- Suellen Hinde; Health reporter (2006-11-26). "Little girl's life of pain". HeraldSun.com.au. Retrieved 2008-07-22.
- Robyn Gobert, Past President of DEBRA Australia Inc. (May 2002). "Times change - A family's story about living with EB". e-bility.com. Retrieved 2008-07-22.
- Gena Brumitt Gruschovnik, DEBRA International Executive Committee. "DEBRA Chile website".
External links
| Classification | |
|---|---|
| External resources |
| Wikimedia Commons has media related to Epidermolysis bullosa. |
- GeneReviews/NCBI/UW/NIH entry on Epidermolysis Bullosa Simplex
- Questions and Answers about Epidermolysis Bullosa - US National Institute of Arthritis and Musculoskeletal and Skin Diseases