ENDOG
Endonuclease G, mitochondrial is an enzyme that in humans is encoded by the ENDOG gene.[5][6] This protein primarily participates in caspase-independent apoptosis via DNA degradation when translocating from the mitochondrion to nucleus under oxidative stress.[7] As a result, EndoG has been implicated in cancer, aging, and neurodegenerative diseases such as Parkinson’s disease (PD). Regulation of its expression levels thus holds potential to treat or ameliorate those conditions.[7][8]



Structure
The enzyme encoded by this gene is a member of the conserved DNA/RNA non-specific ββα-Me-finger nuclease family and possesses a unique site selectivity of poly(dG).poly(dC) sequences in double-stranded DNA. The protein is initially synthesized as an inactive 33-kDa precursor. This precursor is activated by proteolytic cleavage of the mitochondrial targeting sequence, thus producing a mature 28-kDa enzyme that is translocated to the mitochondrial intermembrane space, where it forms an active homodimer.[9][10][11] The H-N-N motif (His-141, Asn-163, Asn-172) is crucial for the protein's catalytic function and substrate specificity, and the His-141 amino acid is necessary for magnesium coordination. The amino acid Asn-251 also appears to be catalytic, and Glu-271 appears to be another magnesium ligand, but both are located far from the H-N-N motif and, thus, their interactions are unclear.[11]
Function
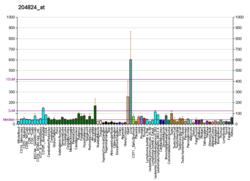
The protein encoded by this gene is a nuclear encoded endonuclease that is localized in the mitochondrial intermembrane space.[6][12] The encoded protein is widely distributed among animals and cleaves DNA at GC tracts. This protein is capable of generating the RNA primers required by DNA polymerase gamma to initiate replication of mitochondrial DNA.[6] In some apoptotic pathways, EndoG is released from the mitochondrion and migrates to the nucleus, where it degrades chromatin with the help of other nuclear proteins.[7][9][11] In one such pathway, caspase-independent apoptosis, the E3 ligase C-terminal of Hsc-70 interacting protein (CHIP), a regulator of EndoG expression, functions as a protective mechanism against oxidative stress. Under normal conditions, EndoG remains bound to Hsp70 and CHIP; however, when undergoing oxidative stress, EndoG dissociates from Hsp70 and CHIP and translocates to the nucleus, where it degrades DNA to effect apoptosis. Therefore, maintaining low levels of EndoG could prevent cell death caused by stress conditions.[13] In epithelial cells, the nuclear localization and proapoptotic function of EndoG leads it to play a role in cell senescence.[10] In addition to DNA degradation, EndoG also stimulates inhibitors of apoptosis proteins (IAPs) to target proteins for proteasomal degradation.[14]
Clinical significance
The Endonuclase G enzyme is an important constituent in apoptotic signaling and oxidative stress, most notably as part of the mitochondrial death pathway and cardiac myocyte apoptosis signaling.[15] Programmed cell death is a distinct genetic and biochemical pathway essential to metazoans. An intact death pathway is required for successful embryonic development and the maintenance of normal tissue homeostasis. Apoptosis has proven to be tightly interwoven with other essential cell pathways. The identification of critical control points in the cell death pathway has yielded fundamental insights for basic biology, as well as provided rational targets for new therapeutics a normal embryologic processes, or during cell injury (such as ischemia-reperfusion injury during heart attacks and strokes) or during developments and processes in cancer, an apoptotic cell undergoes structural changes including cell shrinkage, plasma membrane blebbing, nuclear condensation, and fragmentation of the DNA and nucleus. This is followed by fragmentation into apoptotic bodies that are quickly removed by phagocytes, thereby preventing an inflammatory response.[16] It is a mode of cell death defined by characteristic morphological, biochemical and molecular changes. It was first described as a "shrinkage necrosis", and then this term was replaced by apoptosis to emphasize its role opposite mitosis in tissue kinetics. In later stages of apoptosis the entire cell becomes fragmented, forming a number of plasma membrane-bounded apoptotic bodies which contain nuclear and or cytoplasmic elements. The ultrastructural appearance of necrosis is quite different, the main features being mitochondrial swelling, plasma membrane breakdown and cellular disintegration. Apoptosis occurs in many physiological and pathological processes. It plays an important role during embryonal development as programmed cell death and accompanies a variety of normal involutional processes in which it serves as a mechanism to remove "unwanted" cells.
The BNIP3 pathway involves mitochondrial release and nuclear translocation of the endonuclease G.[17][18] It is not clear, however, that how BNIP3 interacts with mitochondria. It has been shown that BNIP3 interacts with the voltage-dependent anion channel (VDAC) to directly induce mitochondrial release and nuclear translocation of EndonucleaseG. Data has identified VDAC as an interacting partner of BNIP3 and provide direct evidence to support that EndoG is a mediator of the BNIP3 cell death pathway.[19] Most notably, Enodnuclease G is pivotal during oxidative stress by ischemia-reperfusion injury, specifically in the myocardium as part of a heart attack (also known as ischemic heart disease). Ischemic heart disease, which results from an occlusion of one of the major coronary arteries, is currently still the leading cause of morbidity and mortality in western society.[20][21] During ischemia reperfusion, ROS release substantially contribute to the cell damage and death via a direct effect on the cell as well as via apoptotic signals. More recently, Endonuclease G is considered a determinant of cardiac hypertrophy. A link has been established between Endonuclease G and mitochondrial function during cardiac hypertrophy, partly through the effects of Endo G on Mfn2 and Jp2, and revealed a role for Endonuclease G in the crosstalk between the processes controlled by Mfn2 and Jp2 in maladaptive cardiac hypertrophy.[22]
Previous studies reported greater efficacy of anticancer drugs when used in conjunction with high EndoG levels. Thus, regulators of EndoG, such as CHIP, could serve as therapeutic targets for oxidative stress-induced cell death in cancer and aging.[13] Through its association with cell senescence in epithelial cells, EndoG may also contribute to age-related vascular diseases such as arteriosclerosis.[10] Similarly, myonuclear localization of EndoG is correlated with atrophied aging skeletal muscle, leading to increased apoptotic signaling and muscle mass loss. EndoG has also been implicated in Parkinson’s disease (PD), as it induces DNA fragmentation in neurons when translocated from the mitochondria to nuclei. This mechanism involves the kynurenine pathway and the permeability transition pore; as such, targeting molecules in this pathway could prevent EndoG-mediated cell death and effectively help treat PD in patients.[8] Similarly, EndoG knockdown in mice mitigated injurious insults; thus, therapeutic strategies to inhibit or silence EndoG could help protect tissues during injury and disease. So far, two EndoG inhibitors, PNR-3-80 (5-((1-(2-naphthoyl)-5-chloro-1H-indol-3-yl)methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione) and PNR-3-82 (5-((1-(2-naphthoyl)-5-methoxy-1H-indol-3-yl)methylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione, have been tested and confirmed.[9]
Interactions
References
- GRCh38: Ensembl release 89: ENSG00000167136 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000015337 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Tiranti V, Rossi E, Ruiz-Carrillo A, Rossi G, Rocchi M, DiDonato S, Zuffardi O, Zeviani M (Jan 1995). "Chromosomal localization of mitochondrial transcription factor A (TCF6), single-stranded DNA-binding protein (SSBP), and endonuclease G (ENDOG), three human housekeeping genes involved in mitochondrial biogenesis". Genomics. 25 (2): 559–64. doi:10.1016/0888-7543(95)80058-T. PMID 7789991.
- "Entrez Gene: ENDOG endonuclease G".
- Vařecha M, Potěšilová M, Matula P, Kozubek M (Apr 2012). "Endonuclease G interacts with histone H2B and DNA topoisomerase II alpha during apoptosis". Molecular and Cellular Biochemistry. 363 (1–2): 301–7. doi:10.1007/s11010-011-1182-x. PMID 22160858.
- Büttner S, Habernig L, Broeskamp F, Ruli D, Vögtle FN, Vlachos M, Macchi F, Küttner V, Carmona-Gutierrez D, Eisenberg T, Ring J, Markaki M, Taskin AA, Benke S, Ruckenstuhl C, Braun R, Van den Haute C, Bammens T, van der Perren A, Fröhlich KU, Winderickx J, Kroemer G, Baekelandt V, Tavernarakis N, Kovacs GG, Dengjel J, Meisinger C, Sigrist SJ, Madeo F (Nov 2013). "Endonuclease G mediates α-synuclein cytotoxicity during Parkinson's disease". The EMBO Journal. 32 (23): 3041–54. doi:10.1038/emboj.2013.228. PMC 3844953. PMID 24129513.
- Jang DS, Penthala NR, Apostolov EO, Wang X, Crooks PA, Basnakian AG (Feb 2015). "Novel cytoprotective inhibitors for apoptotic endonuclease G". DNA and Cell Biology. 34 (2): 92–100. doi:10.1089/dna.2014.2530. PMC 4308826. PMID 25401220.
- Diener T, Neuhaus M, Koziel R, Micutkova L, Jansen-Dürr P (Aug 2010). "Role of endonuclease G in senescence-associated cell death of human endothelial cells" (PDF). Experimental Gerontology. 45 (7–8): 638–44. doi:10.1016/j.exger.2010.03.002. PMID 20211237.
- Wu SL, Li CC, Chen JC, Chen YJ, Lin CT, Ho TY, Hsiang CY (15 January 2009). "Mutagenesis identifies the critical amino acid residues of human endonuclease G involved in catalysis, magnesium coordination, and substrate specificity". Journal of Biomedical Science. 16: 6. doi:10.1186/1423-0127-16-6. PMC 2653514. PMID 19272175.
- Galluzzi L, Joza N, Tasdemir E, Maiuri MC, Hengartner M, Abrams JM, Tavernarakis N, Penninger J, Madeo F, Kroemer G (Jul 2008). "No death without life: vital functions of apoptotic effectors". Cell Death and Differentiation. 15 (7): 1113–23. doi:10.1038/cdd.2008.28. PMC 2917777. PMID 18309324.
- Lee JS, Seo TW, Yi JH, Shin KS, Yoo SJ (13 June 2013). "CHIP has a protective role against oxidative stress-induced cell death through specific regulation of endonuclease G". Cell Death & Disease. 4 (6): e666. doi:10.1038/cddis.2013.181. PMC 3698548. PMID 23764847.
- Seo TW, Lee JS, Yoo SJ (Sep 2014). "Cellular inhibitor of apoptosis protein 1 ubiquitinates endonuclease G but does not affect endonuclease G-mediated cell death". Biochemical and Biophysical Research Communications. 451 (4): 644–9. doi:10.1016/j.bbrc.2014.08.047. PMID 25139236.
- Danial NN, Korsmeyer SJ (Jan 2004). "Cell death: critical control points". Cell. 116 (2): 205–19. doi:10.1016/S0092-8674(04)00046-7. PMID 14744432.
- Kerr JF, Wyllie AH, Currie AR (Aug 1972). "Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics". British Journal of Cancer. 26 (4): 239–57. doi:10.1038/bjc.1972.33. PMC 2008650. PMID 4561027.
- Zhao ST, Chen M, Li SJ, Zhang MH, Li BX, Das M, Bean JC, Kong JM, Zhu XH, Gao TM (8 September 2009). "Mitochondrial BNIP3 upregulation precedes endonuclease G translocation in hippocampal neuronal death following oxygen-glucose deprivation". BMC Neuroscience. 10: 113. doi:10.1186/1471-2202-10-113. PMC 2749049. PMID 19737385.
- Zhang Z, Yang X, Zhang S, Ma X, Kong J (May 2007). "BNIP3 upregulation and EndoG translocation in delayed neuronal death in stroke and in hypoxia". Stroke: A Journal of Cerebral Circulation. 38 (5): 1606–13. doi:10.1161/STROKEAHA.106.475129. PMID 17379825.
- Zhang X, Bian X, Kong J (2014). "The proapoptotic protein BNIP3 interacts with VDAC to induce mitochondrial release of endonuclease G". PLOS ONE. 9 (12): e113642. doi:10.1371/journal.pone.0113642. PMC 4249980. PMID 25436615.
- Murray CJ, Lopez AD (May 1997). "Alternative projections of mortality and disability by cause 1990-2020: Global Burden of Disease Study". Lancet. 349 (9064): 1498–504. doi:10.1016/S0140-6736(96)07492-2. PMID 9167458.
- Braunwald E, Kloner RA (Nov 1985). "Myocardial reperfusion: a double-edged sword?". The Journal of Clinical Investigation. 76 (5): 1713–9. doi:10.1172/JCI112160. PMC 424191. PMID 4056048.
- Liang X, Ma K, Rao Y, Hong D, Huo Z, Ye Z, Huang M, Zhang X, Zhao Q (Sep 2015). "Characterization of endonuclease G and mitochondria-sarcoplasmic reticulum-related proteins during cardiac hypertrophy". Die Pharmazie. 70 (9): 586–92. PMID 26492643.
Further reading
- Prats E, Noël M, Létourneau J, Tiranti V, Vaqué J, Debón R, Zeviani M, Cornudella L, Ruiz-Carrillo A (Sep 1997). "Characterization and expression of the mouse endonuclease G gene". DNA and Cell Biology. 16 (9): 1111–22. doi:10.1089/dna.1997.16.1111. PMID 9324313.
- Li LY, Luo X, Wang X (Jul 2001). "Endonuclease G is an apoptotic DNase when released from mitochondria". Nature. 412 (6842): 95–9. doi:10.1038/35083620. PMID 11452314.
- Ohsato T, Ishihara N, Muta T, Umeda S, Ikeda S, Mihara K, Hamasaki N, Kang D (Dec 2002). "Mammalian mitochondrial endonuclease G. Digestion of R-loops and localization in intermembrane space". European Journal of Biochemistry / FEBS. 269 (23): 5765–70. doi:10.1046/j.1432-1033.2002.03238.x. PMID 12444964.
- Lemarié A, Lagadic-Gossmann D, Morzadec C, Allain N, Fardel O, Vernhet L (Jun 2004). "Cadmium induces caspase-independent apoptosis in liver Hep3B cells: role for calcium in signaling oxidative stress-related impairment of mitochondria and relocation of endonuclease G and apoptosis-inducing factor". Free Radical Biology & Medicine. 36 (12): 1517–31. doi:10.1016/j.freeradbiomed.2004.03.020. PMID 15182854.
- Singh IN, Goody RJ, Dean C, Ahmad NM, Lutz SE, Knapp PE, Nath A, Hauser KF (Jun 2004). "Apoptotic death of striatal neurons induced by human immunodeficiency virus-1 Tat and gp120: Differential involvement of caspase-3 and endonuclease G". Journal of Neurovirology. 10 (3): 141–51. doi:10.1080/13550280490441103. PMC 4309288. PMID 15204919.
- Kalinowska M, Garncarz W, Pietrowska M, Garrard WT, Widlak P (Aug 2005). "Regulation of the human apoptotic DNase/RNase endonuclease G: involvement of Hsp70 and ATP". Apoptosis. 10 (4): 821–30. doi:10.1007/s10495-005-0410-9. PMID 16133872.
- Basnakian AG, Apostolov EO, Yin X, Abiri SO, Stewart AG, Singh AB, Shah SV (Dec 2006). "Endonuclease G promotes cell death of non-invasive human breast cancer cells". Experimental Cell Research. 312 (20): 4139–49. doi:10.1016/j.yexcr.2006.09.012. PMC 1839947. PMID 17046751.
- Whiteman M, Chu SH, Siau JL, Rose P, Sabapathy K, Schantz JT, Cheung NS, Spencer JP, Armstrong JS (Apr 2007). "The pro-inflammatory oxidant hypochlorous acid induces Bax-dependent mitochondrial permeabilisation and cell death through AIF-/EndoG-dependent pathways". Cellular Signalling. 19 (4): 705–14. doi:10.1016/j.cellsig.2006.08.019. PMID 17107772.
- Varecha M, Amrichová J, Zimmermann M, Ulman V, Lukásová E, Kozubek M (Jul 2007). "Bioinformatic and image analyses of the cellular localization of the apoptotic proteins endonuclease G, AIF, and AMID during apoptosis in human cells". Apoptosis. 12 (7): 1155–71. doi:10.1007/s10495-007-0061-0. PMID 17347867.