Duplex sequencing
Duplex sequencing is a library preparation and analysis method for next-generation sequencing (NGS) platforms that employs random tagging of double stranded DNA to detect mutations with higher accuracy and lower error rate. This method uses degenerate molecular tags in addition to sequencing adapters to recognize reads originating from each strand of DNA. The generated sequencing reads then will be analyzed using two methods: single strand consensus sequences (SSCSs) and Duplex consensus sequences (DCSs) assembly. Duplex sequencing theoretically can detect mutations with frequencies as low as 5 x 10−8 that is more than 10,000 fold higher in accuracy compared to the conventional next-generation sequencing methods.[1][2]
The estimated error rate of standard next-generation sequencing platforms is 10−2 - 10−3 per base call. With this error rate billions of base calls that are produced by NGS will results in millions of errors. The errors are introduced during sample preparation and sequencing such as polymerase chain reaction, sequencing and image analysis errors. While the NGS platforms error rate is admissible to some applications such as detection of clonal variants, it is a major limit for applications that require higher accuracy for detection of low frequency variants such as detection of intra-organismal mosaicism, subclonal variants in genetically heterogeneous cancers or circulating tumor DNA.[3][4][5]
Several library preparation strategies have been developed that increase accuracy of NGS platforms such as molecular barcoding and circular consensus sequencing method.[6][7][8][9] The data generated by these methods the same as NGS platforms originate from single strand of DNA and therefore the errors that are introduced during PCR amplification, tissue processing, DNA extraction, hybridization-capture (where used) or DNA sequencing itself can still be distinguished as a true variant. The duplex sequencing method addresses this problem by taking advantage of complementary nature of two strands of DNA and confirming only variants that are present in both strands of DNA. Because the probability of two complementary errors arising at the same exact same location in both strands is exceedingly low, duplex sequencing increases the accuracy of sequencing significantly.[1][6][8][10]
Experimental workflow
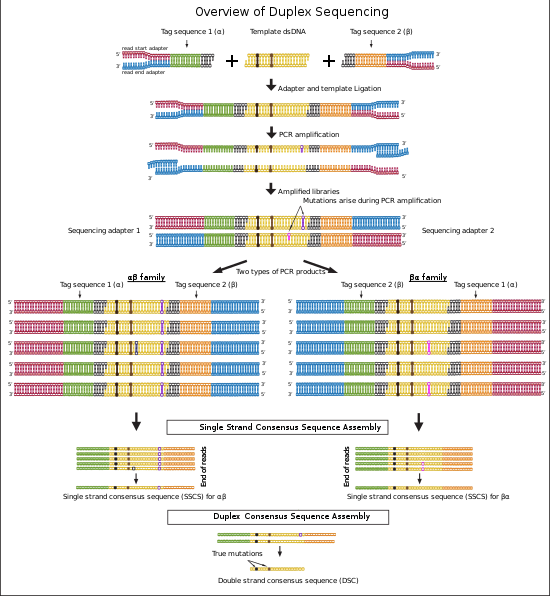
Duplex sequencing tagged adapters can be used in combination with majority of NGS adapters. In the figures and workflow section of this article Illumina sequencing adapters are used as an example in accordance to the original published protocol.[1][2]

Adapter annealing
Two oligonucleotides are used for this step (Figure 1: Adapter oligos). One of the oligonucleotides contains a 12 nucleotide single stranded random tag sequence followed by a fixed 5' nucleotide sequence (Black sequence in figure 1). In this step oligonucleotides are annealed in a complementary region by incubation at the required temporal condition.[1][2]
Adapter synthesis
The adapters that annealed successfully are extended and synthesized by a DNA polymerase to complete a double stranded adapter containing complementary tags (Figure 1).[1][2]
3’-dT-tailing
The extended double stranded adapters are cleaved by HpyCH4III at a specific restriction site located at 3’ side of the tag sequence and will results in a 3’-dT overhang that will be ligated to the 3’-dA overhang on DNA libraries in adapter ligation step (Figure 1).[1][2]
Library preparation
Double stranded DNA is sheared using one of the methods: Sonication, enzymatic digestion or nebulization. Fragments are size selected using Ampure XP beads. Gel-based size selection is not recommended for this method since it can cause melting of DNA double strands and DNA damage as the results of UV exposure. The size selected fragments of DNA are subjected to 3’-end-dA-tailing.[1][2]
Adapter ligation
In this step two tagged adapters are ligated from 3’-dT-tails to 3’-dA-tails on both sides of double stranded DNA library fragments. This process results in double stranded library fragments that contain two random tags (α and β) in each side that are reverse complement of each other (Figure 1 and 2). The "DNA:adapter" ratio is crucial in determining the success of ligation.[1][2]
Insertion of sequencing adapters to tagged libraries
In the last step of duplex sequencing library preparation, Illumina sequencing adapters are added to the tagged double stranded libraries by PCR amplification using primers containing sequencing adapters. During PCR amplification both complementary strands of DNA are amplified and generate two types of PCR products. Product 1 derive from strand 1 which have a unique tag sequence (called α in the figure 2) next to the Illumina adapter 1 and product 2 that have a unique tag (called β in the figure 2) next to illumina adapter 1. (Please note that in each strand, tag α is the reverse complement of tag β and vice versa). The libraries containing duplex tags and Illumina adapters are sequenced using Illumina TruSeq system. Reads that are originating from each single strand of DNA form a group of reads (tag families) that are sharing the same tag. The detected families of reads will be used in next step for analyzing sequencing data.[1][2]
Considerations
Efficiency of adapter ligation
Adapter ligation efficiency is very important in a successful duplex sequencing. Extra amount of libraries or adapters can affect the DNA:adapter balance and therefore result in inefficient ligation and excess amount of primer dimers, respectively. Therefore, it is important to keep the molar concentration of DNA:adapter to the optimal ratio that is 0.05.[2]
Tag family size
Efficiency of duplex sequencing depends on final number of DCSs which is directly related to number of reads in each family (family size). If the family size is too small then the DCS can not be assembled and if too many reads are sharing the same tag the data yield will be low. Family size is determined by the amount of DNA template for PCR amplification and dedicated sequencing lane fraction. The optimal tag family size is between 6 and 12 members. To obtain the optimal family size the amount of DNA template and dedicated sequencing lane fraction needs to be adjusted. The following formula takes into account the most important variables that can affect depth of coverage (N=40DG÷R) where "N" is number of reads, "D" is desired depth of coverage, "G" is size of DNA target in basepair and "R" is final read length.
Computational workflow
Filtering and trimming
Each duplex sequencing read contains a fixed 5-nucleotide sequence (shown in figures in Black color) located upstream of the 12-nucleotide tag sequence. The reads are filtered out if they do not have the expected 5-nucleotide sequence or have more than nine identical or ambiguous bases within each tag. The two 12-nucleotide tags at each end of reads are combined and moved to the read header. Two families of reads are formed that originate from the two strands of DNA. One family contains reads with αβ header originating from strand 1 and the second family contains reads with βα header originating from strand 2 (Figure 2). Then the reads are trimmed by removing the fixed 5 bp sequence and 4 error prone nucleotides located at the sites of ligation and end repair.[1][2] The remaining reads are assembled to consensus sequences using single strand consensus sequences (SSCSs) assembly and duplex consensus sequences (DCSs) assembly.
SSCS assembly
Trimmed sequences from the previous step are aligned to the reference genome using Burrows-Wheeler aligner (BWA) and the unmapped reads are removed. The aligned reads that have the same 24 bp tag sequence and genomic region are detected and grouped together (Family αβ and βα in the figure 2). Each group represents a “tag family”. Tag families with lower than three members are removed from the analysis. To remove errors arise during PCR amplification or sequencing, mutations that are supported by less than 70% of the members (reads) are filtered out from the analysis. Then a consensus sequence is generated for each family using the identical sequences in each position of the remaining reads. The consensus sequence is called single strand consensus sequence (SSCS). The SSCS method increases the NGS accuracy to about 20 fold higher, however this method relies on the sequencing information from single strands of DNA and therefore is sensitive to the errors induced at the first round or before PCR amplification.[1][2]
DCS assembly
The reads from last step are realigned to the reference genome. In this method SSCS family pairs that have complementary tags will be grouped together (Family αβ and βα in figure 2). These reads originate from two complementary strands of DNA. High confidence sequences are selected based on the perfectly matched base calls of each family. The final sequence is called duplex consensus sequence (DCS). True mutations are those that match perfectly between complementary SSCSs. This step filter out remaining errors that raised during first round of PCR amplification or during sample preparation.[1][2]
Advantages
Decreasing error rate of sequencing
High error rate (0.01-0.001) of standard NGS platforms that introduced during sample preparation or sequencing is a major limitation for detection of variants present in small fraction of cells. Due to the duplex tagging system and use of information in both strands of DNA, duplex sequencing has significantly decreased the error rate of sequencing about 10 million fold using both SSCS and DCS method.[1][2][10]
Increasing accuracy of variant calling
It is challenging to identify rare variants accurately using standard NGS methods with the mutations rate of (10−2 - 10−3). Errors that happen early during sample preparation can be detected as rare variants. An example of such errors is C>A/G>T transversion that is detected in low frequencies using deep sequencing or targeted capture data and arise as the result of DNA oxidation during sample preparation.[11] These types of false positive variants are filter out by duplex sequencing method since mutations need to be accurately matched in both strands of DNA to be validated as true mutations. Duplex sequencing can theoretically detect mutations with frequencies as low as 10−8 compare to 10−2 rate of standard NGS methods.[1][2][10]
Applicable to majority of NGS platforms
Another advantage of duplex sequencing is that it can be used in combination with majority of NGS platforms without making significant changes to the standard protocols.
Limitations
Cost
Because duplex sequencing provide a significantly higher sequencing accuracy and uses information in both strands of DNA, this method needs a much higher sequencing depth and therefore is a costly approach. The high cost of duplex sequencing limits its application to targeted and amplicon sequencing at present time and will not be applicable for whole genome sequencing approaches. However, with decreasing cost of NGS, the application of duplex sequencing for larger DNA targets will be more feasible.
Practical application
Duplex sequencing is a new method and its efficiency was studied in limited applications such as detecting point mutations using targeted capture sequencing.[12] More studies need to be performed to expand application and feasibility of duplex sequencing to more complex samples with large number of mutations, indels and copy number variations.
Applications
Detection of variants with low frequencies
Duplex sequencing and the significant increase of sequencing accuracy has important impacts on applications such as detection of rare human genetic variants, detection of subclonal mutations involve in mechanisms of resistance to therapy in genetically heterogeneous cancers, screening variants in circulating tumor DNA as a non-invasive biomarker and prenatal screening for detection of genetic abnormalities in fetus.
Copy number detection
Another suggested application for duplex sequencing is detection of DNA/RNA copy numbers by estimating the relative frequency of variants. A method for counting PCR template molecules with application to next-generation sequencing.[1]
Analysis and software
A list of required tools and packages for SSCS and DCS analysis can be found in software package.
References
- M. W. Schmitt, S. R. Kennedy, J. J. Salk, et al. “Detection of ultra-rare mutations by next-generation sequencing”. Proc. Natl. Acad. Sci., vol. 109 no. 36. 2012. PMID 22853953.
- S. R. Kennedy, M. W. Schmitt, E. J. Fox, B. F. Kohrn, et al. “Detecting ultralow-frequency mutations by Duplex Sequencing”. Nature Protoc., vol. 9 no. 11, 2586-606. 2014. PMID 25299156.
- T. E. Druley, F. L. M. Vallania, D. J. Wegner, et al. “Quantification of rare allelic variants from pooled genomic DNA” Nature Methods, vol. 6, no. 4, pp. 263–265, 2009. PMID 19252504.
- N. McGranahan and C. Swanton. “Biological and Therapeutic Impact of Intratumor Heterogeneity in Cancer Evolution” Cancer Cell, vol. 27, no. 1, pp. 15–26, 2015. PMID 25584892.
- C Bettegowda, M Sausen, RJ Leary, et al. “Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies”. Sci Transl Med, vol. 6, no. 224, p. 224ra24, 2014. PMID 24553385.
- B. E. Miner, R. J. Stöger, A. F. Burden, et al. “Molecular barcodes detect redundancy and contamination in hairpin-bisulfite PCR”. Nucleic Acids Res, vol. 32, no. 17, p. e135, 2004. PMID 15459281.
- M. L. McCloskey, R. Stoger, R. S. Hansen, et al.“Encoding PCR products with batch-stamps and barcodes”, Biochem. Genet., vol. 45, no. 11–12, pp. 761–767, 2007. PMID 17955361.
- D. I. Lou, J. A. Hussmann, R. M. Mcbee, et al. “High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing”. Proc Natl Acad Sci U S A, vol. 110 no. 49, 19872–19877, 2013. PMID 24243955.
- A. Y. Maslov, W. Quispe-Tintaya, T. Gorbacheva, R. R. White, and J. Vijg, “High-throughput sequencing in mutation detection: A new generation of genotoxicity tests?”, Mutat. Res., vol. 776, pp. 136–43, 2015. PMID 25934519.
- E. J. Fox, K. S. Reid-Bayliss, M. J. Emond, et al. “Accuracy of Next Generation Sequencing Platforms”. Next Gener Seq Appl., pp. 1–9, 2015. PMID 25699289.
- M. Costello, T. J. Pugh, T. J. Fennell, et al. “Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation”. Nucleic Acids Res., vol. 41, no. 6, pp. 1–12, 2013. PMID 23303777.
- M. W. Schmitt, E. J. Fox, M. J. Prindle, et al. “Sequencing small genomic targets with high efficiency and extreme accuracy”. Nat Methods, vol. 12, no. 5, pp. 423–425, 2015. PMID 2584963.