Double layer forces
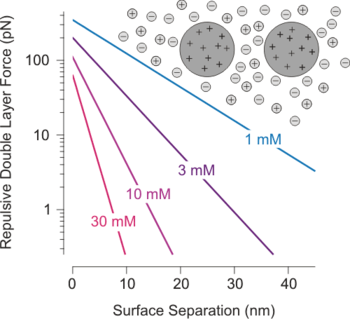
Double layer forces occur between charged objects across liquids, typically water. This force acts over distances that are comparable to the Debye length, which is on the order of one to a few tenths of nanometers. The strength of these forces increases with the magnitude of the surface charge density (or the electrical surface potential). For two similarly charged objects, this force is repulsive and decays exponentially at larger distances, see figure. For unequally charged objects and eventually at shorted distances, these forces may also be attractive. The theory due to Derjaguin, Landau, Verwey, and Overbeek (DLVO) combines such double layer forces together with Van der Waals forces in order to estimate the actual interaction potential between colloidal particles.[1]
An electrical double layer develops near charged surfaces (or another charged objects) in aqueous solutions. Within this double layer, the first layer corresponds to the charged surface. These charges may originate from tightly adsorbed ions, dissociated surface groups, or substituted ions within the crystal lattice. The second layer corresponds to the diffuse layer, which contains the neutralizing charge consisting of accumulated counterions and depleted coions. The resulting potential profile between these two objects leads to differences in the ionic concentrations within the gap between these objects with respect to the bulk solution. These differences generate an osmotic pressure, which generates a force between these objects.
These forces are easily experienced when hands are washed with soap. Adsorbing soap molecules make the skin negatively charged, and the slippery feeling is caused by the strongly repulsive double layer forces.[2] These forces are further relevant in many colloidal or biological systems, and may be responsible for their stability, formation of colloidal crystals, or their rheological properties.
Poisson–Boltzmann model
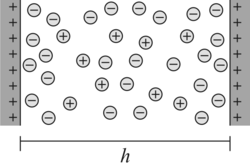
The most popular model to describe the electrical double layer is the Poisson-Boltzmann (PB) model. This model can be equally used to evaluate double layer forces. Let us discuss this model in the case of planar geometry as shown in the figure on the right. In this case, the electrical potential profile ψ(z) near a charged interface will only depend on the position z. The corresponding Poisson's equation reads in SI units

where ρ is the charge density per unit volume, ε0 the dielectric permittivity of the vacuum, and ε the dielectric constant of the liquid. For a symmetric electrolyte consisting of cations and anions having a charge ±q, the charge density can be expressed as

where c± = N±/V are the concentrations of the cations and anions, where N± are their numbers and V the sample volume. These profiles can be related to the electrical potential by considering the fact that the chemical potential of the ions is constant. For both ions, this relation can be written as

where is the reference chemical potential, T the absolute temperature, and k the Boltzmann constant. The reference chemical potential can be eliminated by applying the same equation far away from the surface where the potential is assumed to vanish and concentrations attain the bulk concentration cB. The concentration profiles thus become


where β = 1/(kT). This relation reflects the Boltzmann distribution of the ions with the energy ±qψ. Inserting these relations into the Poisson equation one obtains the PB equation[3]

The potential profile between two plates is normally obtained by solving this equation numerically.
Once the potential profile is known, the force per unit area between the plates expressed as the disjoining pressure Π can be obtained as follows. The starting point is the Gibbs–Duhem relation for a two component system at constant temperature[3]

Introducing the concentrations c± and using the expressions of the chemical potentials μ± given above one finds

The concentration difference can be eliminated with the Poisson equation and the resulting equation can be integrated from infinite separation of the plates to the actual separation h by realizing that

Expressing the concentration profiles in terms of the potential profiles one obtains

From a known electrical potential profile ψ(z) one can calculate the disjoining pressure from this equation at any suitable position z. Alternative derivation of the same relation for disjoining pressure involves the stress tensor.[1]
Debye-Hückel model
| Salt concentration cB (mmol/L) |
Debye length κ−1 (nm) |
|---|---|
| 0.1 | 30 |
| 1 | 10 |
| 10 | 3 |
| 100 | 1 |
When the electric potentials or charge densities are not too high, the PB equation can be simplified to the Debye-Hückel (DH) equation. By expanding the exponential function in the PB equation into a Taylor series, one obtains
- where


The parameter κ−1 is referred to as the Debye length, and some representative values for a monovalent salt in water at 25°C with ε ≃ 80 are given in the table on the right. In non-aqueous solutions, Debye length can be substantially larger than the ones given in the table due to smaller dielectric constants. The DH model represents a good approximation, when the surface potentials are sufficiently low with respect to the limiting values

The numerical value refers to a monovalent salt and 25°C. In practice, the DH approximation remains rather accurate up to surface potentials that are comparable to the limiting values given above. The disjoining pressure can be obtained from the PB equation given above, which can also be simplified to the DH case by expanding into Taylor series. The resulting expression is

The substantial advantage of the DH model over the PB model is that the forces can be obtained analytically. Some of the relevant cases will be discussed below.
Superposition approximation
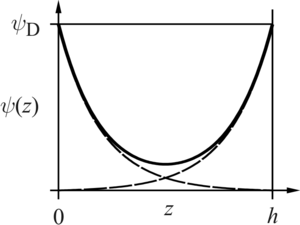
When the surfaces are sufficiently far apart, the potential profiles originating from each individual surface will not be much perturbed by the presence of the other surface. This approximation thus suggests that one can simply add (superpose) the potentials profiles originating from each surface as illustrated the figure. Since the potential profile passes through a minimum at the mid-plane, it is easiest to evaluate the disjoining pressure at the midplane. The solution of the DH equation for an isolated wall reads

where z is the distance from the surface and ψD the surface potential. The potential at the midplane is thus given by twice the value of this potential at a distance z = h/2. The disjoining pressure becomes[1][4]

The electrostatic double layer force decays in an exponential fashion. Due to the screening by the electrolyte, the range of the force is given by the Debye length and its strength by the surface potential (or surface charge density). This approximation turns out to be exact provided the plate-plate separation is large compared to the Debye length and the surface potentials are low.
This result can be simply generalized to highly charged surfaces, but only at larger separations. Even if the potential is large close to the surface, it will be small at larger distances, and can be described by the DH equation. However, in this case one has to replace the actual diffuse layer potential ψD with the effective potential ψeff. Within the PB model, this effective potential can be evaluated analytically, and reads[4]

The superposition approximation can be easily extended to asymmetric systems. Analogous arguments lead to the expression for the disjoining pressure

where the super-scripted quantities refer to properties of the respective surface. At larger distances, oppositely charged surfaces repel and equally charged ones attract.
Charge regulating surfaces
While the superposition approximation is actually exact at larger distances, it is no longer accurate at smaller separations. Solutions of the DH or PB equations in between the plates provide a more accurate picture at these conditions. Let us only discuss the symmetric situation within the DH model here. This discussion will introduce the notion of charge regulation, which suggests that the surface charge (and the surface potential) may vary (or regulate) upon approach.
The DH equation can be solved exactly for two plates.[1][5] The boundary conditions play an important role, and the surface potential and surface charge density and become functions of the surface separation h and they may differ from the corresponding quantities ψD and σ for the isolated surface. When the surface charge remains constant upon approach, one refers to the constant charge (CC) boundary conditions. In this case, the diffuse layer potential will increase upon approach. On the other hand, when the surface potential is kept constant, one refers to constant potential (CP) boundary condition. In this case, the surface charge density decreases upon approach. Such decrease of charge can be caused by adsorption of desorption of charged ions from the surface. Such variation of adsorbed species upon approach has also been referred to as proximal adsorption.[6] The ability of the surface to regulate its charge can be quantified by the regulation parameter



where CD = ε0 ε κ is the diffuse layer capacitance and CI the inner (or regulation) capacitance. The CC conditions are found when p = 1 while the CP conditions for p = 0. The realistic case will be typically situated in between. By solving the DH equation one can show that diffuse layer potential varies upon approach as

while the surface charged density obey a similar relation

The swelling pressure can be found by inserting the exact solution of the DH equation into the expressions above and one finds

Repulsion is strongest for the CC conditions (p = 1) while it is weaker for the CP conditions (p = 0). The result of the superposition approximation is always recovered at larger distances but also for p = 1/2 at all distances. The latter fact explains why the superposition approximation can be very accurate even at small separations. Surfaces regulate their charge and not infrequently the actual regulation parameter is not far away from 1/2. The situation is exemplified in the figure below. From stability considerations one can show that p < 1 and that this parameter may also becomes negative. These results can be extended to asymmetric case in a straightforward way.[5]
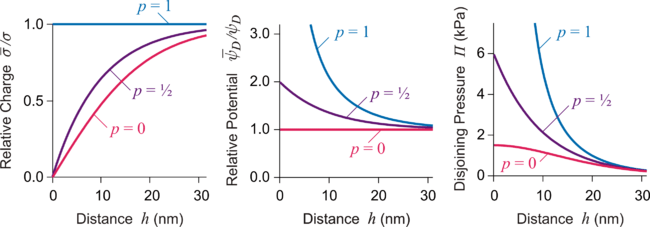
When surface potentials are replaced by effective potentials, this simple DH picture is applicable for more highly charged surfaces at sufficiently larger distances. At shorter distances, however, one may enter the PB regime and the regulation parameter may not remain constant. In this case, one must solve the PB equation together with an appropriate model of the surface charging process. It was demonstrated experimentally that charge regulation effects can become very important in asymmetric systems.[7]
Extensions to other geometries
Interactions between various objects were studied within the DH and PB models by many researchers. Some of the relevant results are summarized in the following.
Non-planar geometries: Objects of other than planar geometries can be treated within the Derjaguin approximation, provided their size is substantially larger than the Debye length. This approximation has been used to estimate the force between two charged colloidal particles as shown in the first figure of this article. The exponential nature of these repulsive forces and the fact that its range is given by the Debye length was confirmed experimentally by direct force measurements, including surface forces apparatus,[3][8] colloidal probe technique,[7][9] or optical tweezers.[10][11] The interaction free energy involving two spherical particles within the DH approximation follows the Yukawa or screened Coulomb potential[4][12]

where r is the center-to-center distance, Q is the particle charge, and a the particle radius. This expression is based on the superposition approximation and is only valid at large separations. This equation can be extended to more highly charged particles by reinterpreting the charge Q as an effective charge. To address the interactions in other situation, one must resort to numerical solutions of the DH or PB equation.
Non-uniform or patchy charge distribution: Interaction between surfaces with non-uniform and periodic charge distribution has been analyzed within the DH approximation.[13][14] Such surfaces are referred to have a mosaic or patch-charge distribution. One important conclusion from these studies is that there is an additional attractive electrostatic contribution, which also decays exponentially. When the non-uniformities are arranged in a quadratic lattice with spacing b, the decay length q−1 of this additional attraction can be expressed as

At high salt levels, this attraction is screened as the interaction between uniformly charged surfaces. At lower salt levels, however, the range of this attraction is related to the characteristic size of the surface charge heterogeneities.
Three-body forces: The interactions between weakly charged objects are pair-wise additive due to the linear nature of the DH approximation. On the PB level, however, attractive three-body forces are present.[11] The interaction free energy between three objects 1, 2, and 3 can be expressed as

where Fij are the pair free energies and ΔF123 is the non-additive three-body contribution. These three-body contributions were found to be attractive on the PB level, meaning that three charged objects repel less strongly than what one would expect on the basis of pair-wise interactions alone.
Beyond Poisson-Boltzmann approximation
More accurate description of double layer interactions can be put forward on the primitive model. This model treats the electrostatic and hard-core interactions between all individual ions explicitly. However, it includes the solvent only in a "primitive" way, namely as a dielectric continuum. This model was studied in much detail in the theoretical community.[12][15][16][17] Explicit expressions for the forces are mostly not available, but they are accessible with computer simulations, integral equations, or density functional theories.
The important finding from these studies is that the PB description represents only a mean-field approximation. This approximation is excellent in the so-called weak coupling regime, that is for monovalent electrolytes and weakly charged surfaces. However, this description breaks down in the strong coupling regime, which may be encountered for multivalent electrolytes, highly charged systems, or non-aqueous solvents.[17] In the strong coupling regime, the ions are strongly correlated, meaning that each ion has an exclusion hole around itself. These correlations lead to strong ion adsorption to charged surfaces, which may lead to charge reversal and crystallization of these ions on the surface. These correlations may also induce attractive forces. The range of these forces is typically below 1 nm.
Like-charge attraction controversy
Around 1990, theoretical and experimental evidence has emerged that forces between charged particles suspended in dilute solutions of monovalent electrolytes might be attractive at larger distances.[18][19] This evidence is in contradiction with the PB theory discussed above, which always predicts repulsive interactions in these situations. The theoretical treatment leading to these conclusions was strongly criticized.[20][21] The experimental findings were mostly based on video-microscopy, but the underlying data analysis was questioned concerning the role of impurities, appropriateness of image processing techniques,[10] and the role of hydrodynamic interactions.[22]
While the community remains skeptical regarding the existence of effective attractions between like-charged species, recent computer molecular dynamics simulations with an explicit description of the solvent have demonstrated that the solvent plays an important role for the structure of charged species in solution while PB and the primitive model do not account for most of these effects.[23] Specifically, the solvent plays a key role in the charge localization of the diffuse ions in ion-rich domains that bring charged species closer together. Based on this idea, simulations have explained experimental trends such as the disappearance of a scattering peak in salt-free polyelectrolyte solutions that PB and primitive model approaches fail to explain.[24]
Relevance
Double layer interactions are relevant in a wide number of phenomena.[4] These forces are responsible for swelling of clays. They may also be responsible for the stabilization of colloidal suspension and will prevent particle aggregation of highly charged colloidal particles in aqueous suspensions. At low salt concentrations, the repulsive double layer forces can become rather long-ranged, and may lead to structuring of colloidal suspensions and eventually to formation of colloidal crystals. Such repulsive forces may further induce blocking of surfaces during particle deposition. Double layer interactions are equally relevant for surfactant aggregates, and may be responsible to the stabilization of cubic phases made of spheroidal micelles or lamellar phases consisting of surfactant or lipid bilayers.
See also
References
- W. B. Russel, D. A. Saville, W. R. Schowalter, Colloidal Dispersions. Cambridge University Press: Cambridge, 1989.
- Idson, Bernard (1967). "Adsorption to Skin and Hair". Journal of the Society of Cosmetic Chemists. 18: 91–103 – via Society of Cosmetic Chemists.
- J. Israelachvili, Intermolecular and Surface Forces. Academic Press: London, 1992.
- D. F. Evans, H. Wennerstöm, The Colloidal Domain, John Wiley-VCH, New York, 1999.
- Carnie, Steven L.; Chan, Derek Y.C. (1993). "Interaction Free Energy between Plates with Charge Regulation: A Linearized Model". Journal of Colloid and Interface Science. Elsevier BV. 161 (1): 260–264. doi:10.1006/jcis.1993.1464. ISSN 0021-9797.
- Subramanian, Vivek; Ducker, William (2001). "Proximal Adsorption of Cationic Surfactant on Silica at Equilibrium". The Journal of Physical Chemistry B. American Chemical Society (ACS). 105 (7): 1389–1402. doi:10.1021/jp003168f. ISSN 1520-6106.
- Popa, Ionel; Sinha, Prashant; Finessi, Marco; Maroni, Plinio; Papastavrou, Georg; Borkovec, Michal (2010-06-02). "Importance of Charge Regulation in Attractive Double-Layer Forces between Dissimilar Surfaces". Physical Review Letters. American Physical Society (APS). 104 (22): 228301. doi:10.1103/physrevlett.104.228301. ISSN 0031-9007.
- Pashley, R.M (1981). "DLVO and hydration forces between mica surfaces in Li+, Na+, K+, and Cs+ electrolyte solutions: A correlation of double-layer and hydration forces with surface cation exchange properties". Journal of Colloid and Interface Science. Elsevier BV. 83 (2): 531–546. doi:10.1016/0021-9797(81)90348-9. ISSN 0021-9797.
- Kane, Victoria; Mulvaney, Paul (1998). "Double-Layer Interactions between Self-Assembled Monolayers of ω-Mercaptoundecanoic Acid on Gold Surfaces". Langmuir. American Chemical Society (ACS). 14 (12): 3303–3311. doi:10.1021/la971296y. ISSN 0743-7463.
- Gutsche, C.; Keyser, U. F.; Kegler, K.; Kremer, F.; Linse, P. (2007-09-25). "Forces between single pairs of charged colloids in aqueous salt solutions". Physical Review E. American Physical Society (APS). 76 (3): 031403. doi:10.1103/physreve.76.031403. ISSN 1539-3755.
- Dobnikar, Jure; Brunner, Matthias; von Grünberg, Hans-Hennig; Bechinger, Clemens (2004-03-10). "Three-body interactions in colloidal systems". Physical Review E. American Physical Society (APS). 69 (3): 031402. doi:10.1103/physreve.69.031402. ISSN 1539-3755.
- Levin, Yan (2002-09-26). "Electrostatic correlations: from plasma to biology". Reports on Progress in Physics. IOP Publishing. 65 (11): 1577–1632. arXiv:cond-mat/0207086. doi:10.1088/0034-4885/65/11/201. ISSN 0034-4885.
- P. Richmond, J. Chem. Soc. Farad. Trans. II 70 (1974) 1066-1073.
- Miklavic, S. J.; Chan, D. Y. C.; White, L. R.; Healy, T. W. (1994). "Double Layer Forces between Heterogeneous Charged Surfaces". The Journal of Physical Chemistry. American Chemical Society (ACS). 98 (36): 9022–9032. doi:10.1021/j100087a034. ISSN 0022-3654.
- Guldbrand, Lars; Jönsson, Bo; Wennerström, Håkan; Linse, Per (1984). "Electrical double layer forces. A Monte Carlo study". The Journal of Chemical Physics. AIP Publishing. 80 (5): 2221–2228. doi:10.1063/1.446912. ISSN 0021-9606.
- Kjellander, Roland; Marcělja, Stjepan (1984). "Correlation and image charge effects in electric double layers". Chemical Physics Letters. Elsevier BV. 112 (1): 49–53. doi:10.1016/0009-2614(84)87039-6. ISSN 0009-2614.
- Kjellander, Roland; Åkesson, Torbjörn; Jönsson, Bo; Marčelja, Stjepan (1992-07-15). "Double layer interactions in mono‐ and divalent electrolytes: A comparison of the anisotropic HNC theory and Monte Carlo simulations". The Journal of Chemical Physics. AIP Publishing. 97 (2): 1424–1431. doi:10.1063/1.463218. ISSN 0021-9606.
- Sogami, Ikuo; Ise, Norio (1984-12-20). "On the electrostatic interaction in macroionic solutions". The Journal of Chemical Physics. AIP Publishing. 81 (12): 6320–6332. doi:10.1063/1.447541. ISSN 0021-9606.
- Crocker, John C.; Grier, David G. (1996-08-26). "When Like Charges Attract: The Effects of Geometrical Confinement on Long-Range Colloidal Interactions". Physical Review Letters. American Physical Society (APS). 77 (9): 1897–1900. doi:10.1103/physrevlett.77.1897. ISSN 0031-9007.
- Overbeek, J. Theodoor G. (1987-10-15). "On the electrostatic interaction in macroionic solutions and suspensions". The Journal of Chemical Physics. AIP Publishing. 87 (8): 4406–4408. doi:10.1063/1.452851. ISSN 0021-9606.
- Wu, J.; Bratko, D.; Prausnitz, J. M. (1998-12-22). "Interaction between like-charged colloidal spheres in electrolyte solutions". Proceedings of the National Academy of Sciences USA. Proceedings of the National Academy of Sciences. 95 (26): 15169–15172. doi:10.1073/pnas.95.26.15169. ISSN 0027-8424.
- Squires, Todd M.; Brenner, Michael P. (2000-12-04). "Like-Charge Attraction and Hydrodynamic Interaction". Physical Review Letters. American Physical Society (APS). 85 (23): 4976–4979. arXiv:cond-mat/0003195. doi:10.1103/physrevlett.85.4976. ISSN 0031-9007.
- Chremos, A.; Douglas, J. (2019-03-08). "Influence of solvation on the structure of highly charged nanoparticles in salt-free solutions". Polymer. Elsevier. 170: 107–112. doi:10.1016/j.polymer.2019.03.005.
- Chremos, A.; Horkay, F. (2020-07-27). "Disappearance of the polyelectrolyte peak in salt-free solutions". Phys. Rev. E. American Physical Society (APS). 102: 012611. doi:10.1103/PhysRevE.102.012611.