Development of the cerebral cortex
Corticogenesis is the process in which the cerebral cortex of the brain is formed during the development of the nervous system. The cortex is the outer layer of the brain and is composed of up to six layers. Neurons formed in the ventricular zone migrate to their final locations in one of the six layers of the cortex.[1] The process occurs from embryonic day 10 to 17 in mice and between gestational weeks seven to 18 in humans.[2]
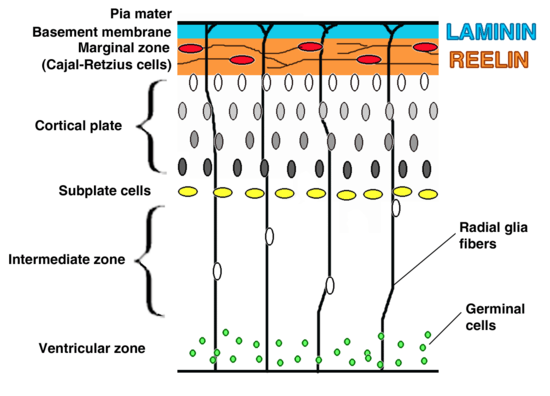
Cortical plates and zones
Plates
The preplate is the first stage in corticogenesis prior to the development of the cortical plate. The preplate is located between the pia and the ventricular zone. According to current knowledge, the preplate contains the first-born or pioneer neurons. These neurons are mainly thought to be Cajal-Retzius cells. The preplate also contains the predecessor to the subplate, which is sometimes referred to as a layer. As the cortical plate appears, the preplate separates into two components. The Cajal-Retzius cells go into the marginal zone, above the cortical plate, while the subplate moves to below the 6 cortical layers.[1] It is during this transition from preplate to cortical plate when many malformations may arise.
The cortical plate is the final plate formed in corticogenesis. It includes the cortex layers two through six.[1]
The subplate is located beneath the cortical plate. It is named for both its location relative to the cortical plate and for the time frame in which it is created. While cortical plate matures, the cells located in the subplate establish connections with neurons that have not yet moved to their destination layer within the cortical plate. Pioneer cells are also present in the subplate and work to create fibers and synapses within the plate.[1]
Zones
The intermediate zone is located between the ventricular zone and the cortical plate. The white matter in this area is where neurons, that are created in the ventricular zone, migrate through in order to reach the cortical plate.[1] This zone is only present during corticogenesis and eventually transforms into adult white matter.
The ventricular and subventricular zones exist below the intermediate zone and communicate to other zones through cell signalling, also creating neurons destined to migrate to other areas in the cortex.[1][3]
The marginal zone, along with the cortical zone, make up the 6 layers that form the cortex. This zone is the predecessor for layer 1 of the cortex. Astrocytes form an outer limiting membrane to interact with the pia. In humans it has been found that the cells here also form a subpial layer.[1] Cajal-Retzius cells are also present in this zone and release reelin along the radial axis, a key to proper neuronal migration during corticogenesis.[4]
Formation of layers
The cerebral cortex is divided into layers. Each layer is formed by radial glial cells located in the ventricular zone or subventricular zone, and then migrate to their final destination.[5]
Layer I
Layer I, the molecular layer, is the first cortical layer produced during neurogenesis at mouse E10.5 to E12.5.[4] Of the six layers found within the neocortex, layer I is the most superficial composed of Cajal–Retzius cells and pyramidal cells.[5] This layer is unique in the aspect that these cells migrate to the outer edge of the cortex opposed to the migration experienced by the other 5 layers. Layer one is also characterized by expression of reelin, transcription factor T-box brain 1, and cortical migratory neuronal marker.[1]
Layers 2 and 3
The second and third layers, or the External Granular layer and External Pyramidal layer respectively, are formed around mouse E13.5 to E16. These layers are the last to form during corticogenesis and include pyramidal neurons, astrocytes, Stellates, and radial glial cells. The pyramidal and stellate neurons express SATB2 and CUX1. SATB2 and CUX1 are DNA binding proteins involved in determining the fate of cortical cells.[5]
Layers 4, 5 and 6
The fourth, fifth and sixth layers, or the Internal Granular layer, Internal Pyramidal layer, and Polymorphic or Multiform layer respectively, are formed during mouse E11.5 to E14.5. Included in these layers are stellates, radial glia, and pyramidal neurons. Layer six is adjacent to the ventricular zone. During the production of these layers, transcription factors TBR1 and OTX1 are expressed along with CTIP2, or corticoneuronal zinc finger protein.[5]
Neuronal migration
Neuronal migration plays significant role in corticogenesis. Throughout the process of creating the six cortical layers, all the neurons and cells migrate from the ventricular zone, through the subplate, and come to rest at their appropriate layer of the cortex. Neuronal migration is generally subdivided into radial migration, tangential migration and multipolar migration.[1] The migration of subcortical brain functions to the cortex is known as corticalization.[6]
Cell signaling
Appropriate formation of the cerebral cortex relies heavily on a densely intertwined network of multiple signaling pathways and distinct signaling molecules. While the majority of the process remains to be understood, some signals and pathways have been carefully unraveled in an effort to gain full knowledge of the mechanisms that control corticogenesis.
Reelin-DAB1 pathway
The Reelin-DAB1 pathway is a well-defined pathway involved in corticogenesis.[7] Cajal-Retzius cells located in the marginal zone secrete reelin to start the cascade. Reelin is able to interact with specific neurons in the cortical plate and direct these neurons to their proper locations. It is thought that the result downstream from this signalling could influence the cytoskeleton. Reelin is secreted only by the Cajal-Retzius cells located in the marginal zone, and its receptors are confined to the cortical plate. This segregation could be used to understand the actions of Reelin.[1]
DAB1 is a regulator protein downstream of the reelin receptors. This protein is located inside cells residing in the ventricular zone, displaying highest concentrations in migrating pyramidal cells. When either reelin or DAB1 are inactivated in mice, the resulting phenotypes are the same. In this case, the neurons are unable to migrate properly through the cortical plate. It does not affect the proliferation of neurons and in the wild does not seem to have detrimental effects on memory or learning.[1][3]
Sonic hedgehog
Knocking out the Sonic hedgehog, or Shh, has been shown to severely affect corticogenesis in the genetically modified mice. The ventral and dorsal sides of the cerebrum are affected as Shh expresses the transcription factors to Nkx2 which is important in patterning the cortex. Shh is also important to corticogenesis as it affects cell proliferation and differentiation, helping neuronal progenitor cells in fate determination.[8]
Bmp-7
Bone morphogenetic protein 7 (Bmp-7), is an important regulator in corticogenesis, though it is not understood whether it promotes or inhibits neurogenesis. Bmp-7 can be detected in the ventricular zone and is secreted into cerebrospinal fluid (CSF). The CSF is an area to promote neurogenesis and it is believed that the synergy between Bmp-7 and other regulators promote cell division along with homeostasis.[9]
Other bone morphogenetic proteins are also known to impact corticogenesis. Bmp2, 4, 5, and 6 are expressed during the process and can compensate for one another. For example, if Bmp-4 was absent from corticogenesis, very little would change in the cortex phenotype, due to the other Bmps helping accomplish the tasks of Bmp-4. However, Bmp-7 is the only Bmp that promotes radial glia survival and therefore considered more important.[9]
Cdk5-p35 pathway
Cdk5 has a pathway parallel to the Reelin-DAB1. This pathway affects the neuronal positioning, and results in similar malformations when absent as the Reelin or DAB1 malformations except that migration is affected at an earlier stage on the cortical plate. Cdk5/p35 pathway is also responsible for actin and microtubule dynamics involved in neuronal migration.[1]
Cyclin-dependent kinase inhibitor 1C, or p57, also affects corticogenesis. It has been shown the p57 induces cells to exit from the cell cycle and begin differentiation, but it is dependent on Cdks. p57 is able to induce neuronal progenitor cells to start differentiating into highly specialized neurons in the cortex. However, the mechanism by which p57 is able to affect such control is not yet known.[10]
Other signals
Besides the ones listed above, there are several more signals that affect corticogenesis. Cnr1 is a g protein receptor that is widely expressed throughout the brain, and in interneurons. In knockout mice, the cortex exhibited decreased immunoreactivity. Nrp1, Robo1, and Robo2 have also been shown to be present and important in the development of interneurons. Cdh8 is known to be expressed in the intermediate and subventricular zone, though not in specific neurons in that area, and it is suggested to regulate fiber releasing.[3]
Disorders
Lissencephaly
Lissencephaly, or 'smooth brain', is a disorder in which the brain does not properly form the gyri and sulci as a result from neuronal migration and cortical folding. This disorder can also result in epilepsy and cognitive impariment.[11] Type 1 lissencephaly is due to an error in migration. LISI, also known as PAFAH1B, is expressed in both dividing and migrating cells found in the brain. When LIS1 is deleted, lissencephaly occurs.[1]
LIS1 is thought to have several important roles in the creation of the cortex. Since LIS1 is similar to the nuclear distribution protein F (nudF), they are thought to work similarly. The nud family is known to be a factor in nuclear translocation, or moving the nuclei of daughter cells after cell division has occurred.[11] By relation, it is thought that LIS1 is a factor in neuronal migration. LIS1 is also considered to be a factor in controlling dynein, a motor protein that affects intercellular movement such as protein sorting and the process of cell division.[1]
Another protein that contributes to a lissencephaly disorder is DCX, or Doublecortin. DCX is a microtubule associated protein that is responsible for double cortex malformations.[1] DCX is found in the second layer of the cortex, and in fact is still present in immature neurons of an adult cortex.[12] It is thought that DCX affects neuronal migration through affecting the microtubule dynamics. Since DCX malformations results as a similar phenotype as with LIS1 malformations, it is thought they interact with one another on a cellular level. However, it is not yet known how this occurs.[1]
Tsc1 knockout
TSC, or tuberous sclerosis, is an autosomal dominate disorder. TSC1 or TSC2 inactivation can cause TSC and the associated tumors in the brain. When inactivation of TSC1 is present during corticogenesis, malformations of cortical tubers, or abnormal benign tissue growth, along with white matter nodes would form in mice. This replicates the effect TSC is found to have in humans afflicted with TSC. In the mice there would be a lack of GFAP in astrocytes however astrogliosis would not occur like in the human TSC.[13]
Human Cortex Malformation (Overfolding)
The sodium channel SCN3A has been implicated in cortical malformations .[14]
Recapitulation
Recapitulation of corticogenesis in both human and mouse embryos have been accomplished with a three dimensional culture using embryonic stem cells (ESC). Recapitulation is the theory in which an organism passes through embryonic development in stages similar to evolution of that organism. By carefully using embryo body intermediates and cultured in a serum free environment cortical progenitors form in a space and time related pattern similar to in vivo corticogenesis. Using immunocytochemical analysis on mouse neural stem cells derived from ESCs, after 6 days there was evidence of neuronal differentiation.[5] The recapitulation ability only follows after the knowledge of spatial and temporal patterns have been identified, along with giving the knowledge that corticogenesis can occur without input from the brain.[15]
References
- Meyer, G. (2007). Genetic Control of Neuronal Migrations in Human Cortical Development(Advances in Anatomy, Embryology and Cell Biology). F. F. Beck, Melbourne, F. Clascá, Madrid, M. Frotscher, Freiburg, D. E. Haines, Jackson, H-W. Korf, Frankfurt, E.Marani, Enschede, R. Putz, München, Y. Sano, Kyoto, T. H. Schiebler, Würzburg & K. Zilles, Düsseldorf (Eds). New York, NY:Springer.
- , Haydar TF, Blue ME, Molliver ME, Krueger BK, Yarowsky PJ. Consequences of trisomy 16 for mouse brain development: corticogenesis in a model of Down syndrome. J Neurosci. 1996 Oct 1;16(19):6175-82. PubMed PMID 8815899.
- Antypa, M., Faux, C., Eichele, G., Parnavelas, J. G., & Andrews, W. D. (2011). Differential gene expression in migratory streams of cortical interneurons. European Journal of Neuroscience, 34(10), 1584-1594. doi: 10.1111/j.1460-9568.2011.07896.x
- Kwon, H. J., Ma, S., & Huang, Z. (2011). Radial glia regulate Cajal-Retzius cell positioning in the early embryonic cerebral cortex. Developmental Biology, 351(1), 25-34. doi: 10.1016/j.ydbio.2010.12.026
- Germain, N., Banda, E., & Grabel, L. (2010). Embryonic Stem Cell Neurogenesis and Neural Specification. Journal of Cellular Biochemistry, 111(3), 535-542. doi: 10.1002/jcb.22747
- "corticalization". The Free Dictionary.
- "Impaired Reelin-Dab1 Signalling contributes to Neuronal Migration Deficits". doi:10.1016/j.celrep.2015.07.013. PMC 4536164. Cite journal requires
|journal=(help) - Komada, M. (2012). Sonic hedgehog signaling coordinates the proliferation and differentiation of neural stem/progenitor cells by regulating cell cycle kinetics during development of the neocortex. Congenital Anomalies, 52(2), 72-77. doi: 10.1111/j.1741-4520.2012.00368.x
- Segklia, A., Seuntjens, E., Elkouris, M., Tsalavos, S., Stappers, E., Mitsiadis, T. A., . . . Graf, D. (2012). Bmp7 Regulates the Survival, Proliferation, and Neurogenic Properties of Neural Progenitor Cells during Corticogenesis in the Mouse. PLoS ONE, 7(3). doi: 10.1371/journal.pone.0034088
- Tury, A., Mairet-Coello, G., & DiCicco-Bloom, E. (2011). The Cyclin-Dependent Kinase Inhibitor p57(Kip2) Regulates Cell Cycle Exit, Differentiation, and Migration of Embryonic Cerebral Cortical Precursors. Cerebral Cortex, 21(8), 1840-1856. doi: 10.1093/cercor/bhq254
- Toba, S., & Hirotsune, S. (2012). A unique role of dynein and nud family proteins in corticogenesis. Neuropathology, 32(4), 432-439. doi: 10.1111/j.1440-1789.2012.01301.x
- Zhang, M. Q., Wang, H., & Xiong, K. (2011). Is the neocortex a novel reservoir for adult mammalian neurogenesis? Neural Regeneration Research, 6(17), 1334-1341. doi: 10.3969/j.issn.1673-5374.2011.17.009
- Feliciano, D. M., Su, T., Lopez, J., Platel, J. C., & Bordey, A. (2011). Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. Journal of Clinical Investigation, 121(4), 1596-1607. doi: 10.1172/jci44909
- Smith, RS; Kenny, CJ; Ganesh, V; Jang, A; Borges-Monroy, R; Partlow, JN; Hill, RS; Shin, T; Chen, AY; Doan, RN; Anttonen, AK; Ignatius, J; Medne, L; Bönnemann, CG; Hecht, JL; Salonen, O; Barkovich, AJ; Poduri, A; Wilke, M; de Wit, MCY; Mancini, GMS; Sztriha, L; Im, K; Amrom, D; Andermann, E; Paetau, R; Lehesjoki, AE; Walsh, CA; Lehtinen, MK (5 September 2018). "Sodium Channel SCN3A (NaV1.3) Regulation of Human Cerebral Cortical Folding and Oral Motor Development". Neuron. 99 (5): 905–913.e7. doi:10.1016/j.neuron.2018.07.052. PMC 6226006. PMID 30146301.
- Gaspard N, Bouschet T, Hourex R, Dimidschstein J, Naeije G, van den Ameele J, Espuny-Camacho I, Herpoel A, Passante L, Schiffmann SN, Gaillard A, Vanderhargen P. (2008). An Intrinsic mechanism of corticogenesis from embryonic stem cells. Nature, 455:351-357.