Central nervous system cavernous hemangioma
Cerebral Cavernous Malformation (CCM) is a cavernous hemangioma that arises in the central nervous system (CNS). It can be considered to be a variant of hemangioma, and is characterized by grossly large dilated blood vessels and large vascular channels, less well circumscribed, and more involved with deep structures, with a single layer of endothelium and an absence of neuronal tissue within the lesions. These thinly walled vessels resemble sinusoidal cavities filled with stagnant blood. Blood vessels in patients with cerebral cavernous malformations (CCM) can range from a few millimeters to several centimeters in diameter. Most lesions occur in the brain, but any organ may be involved.[1]
| Cerebral Cavernous Malformation (CCM) | |
|---|---|
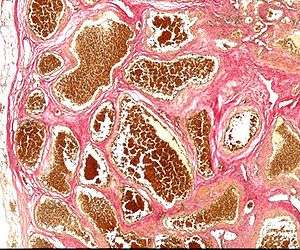 | |
| Histology of a cavernous hemangioma | |
| Specialty | Oncology |
Symptoms
Clinical symptoms of CNS origin include recurrent headaches, focal neurological deficits, hemorrhagic stroke, and seizures, but CCM can also be asymptomatic. The nature and severity of the symptoms depend on the lesion's location.
CCMs and venous angiomas
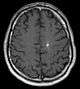
In up to 30% there is a coincidence of CCM with a venous angioma, also known as a developmental venous anomaly (DVA). These lesions appear either as enhancing linear blood vessels or caput medusae, a radial orientation of small vessels that resemble the hair of Medusa from Greek mythology. These lesions are thought to represent developmental anomalies of normal venous drainage. These lesions should not be removed, as venous infarcts have been reported. When found in association with a CCM that needs resection, great care should be taken not to disrupt the angioma.
Genetics
Familial forms of CCM occur at three known genetic loci. The gene for CCM1 encodes KRIT1 (krev interaction trapped 1),[2][3] and has been found to bind to ICAP1alpha (integrin cytoplasmic domain associated protein alpha),[4] a beta1 integrin associated protein. A particular mutation in CCM1 (the Q455X mutation), accounts for a cluster of cases in the Southwestern United States.[5] This cluster, particularly in northern New Mexico, is an example of the founder effect; it has been traced back to early Spanish settlers.[6]
The gene for CCM2 encodes a protein named malcavernin that contains a phosphotyrosine (PTB) binding domain.[7] The exact biological function of CCM2 is not clear. Recently, it has been shown that CCM1 and CCM2 proteins as well as ICAP1alpha form a macromolecular complex in the cell. In addition, it appears that CCM2 protein may function as a scaffolding protein for MAP kinases that are essential in p38 activation responding to osmotic stress including MEKK3 and MKK3.[8] It also binds to Rac and actin. Therefore, CCM2 protein is also called OSM (osmosensing scaffold for MEKK3).
The CCM3 gene is the most recently identified CCM gene . CCM3 is known as PDCD10 (programmed cell death 10), which was initially identified as a gene that is up-regulated during the induction of apoptosis (cell death) in TF-1, a human myeloid cell line.[9] The precise role of the PDCD10 protein in the CCM pathway is not clear. It is recently shown that PDCD10 forms a complex with CCM1 protein (KRIT1) and CCM2 protein (OSM). PDCD10 interacts directly with OSM independent of KRIT1-OSM interaction. Research is ongoing to determine the function and properties of all three CCM gene products as well as the reaction pathways in which they are involved.
Evidence suggests that a fourth gene, CCM4, may also cause CCM.[10] Mutations in these genes account for 70 to 80 percent of all cases of cerebral cavernous malformations. The remaining 20 to 30 percent of cases may be due to other, still unidentified, genes. Recently it has been shown that the deletion of CDC42 in endothelial cells elicits cerebral vascular malformations, suggesting that it may be a fourth gene involved in CCM pathology[11].
Diagnosis
Diagnosis is generally made by magnetic resonance imaging (MRI), particularly using a specific imaging technique known as a gradient-echo sequence MRI, which can unmask small or punctate lesions that may otherwise remain undetected. These lesions are also more conspicuous on FLAIR imaging compared to standard T2 weighing. FLAIR imaging is different from gradient sequences. Rather, it is similar to T2 weighing but suppresses free-flowing fluid signal. Sometimes quiescent CCMs can be revealed as incidental findings during MRI exams ordered for other reasons. Many cavernous hemangiomas are detected "accidentally" during MRIs searching for other pathologies. These "incidentalomas" are generally asymptomatic. In the case of hemorrhage, however, a CT scan is more efficient at showing new blood than an MRI, and when brain hemorrhage is suspected, a CT scan may be ordered first, followed by an MRI to confirm the type of lesion that has bled.
Sometimes the lesion appearance imaged by MRI remains inconclusive. Consequently, neurosurgeons will order a cerebral angiogram or magnetic resonance angiogram (MRA). Since CCMs are low flow lesions (they are hooked into the venous side of the circulatory system), they will be angiographically occult (invisible). If a lesion is discernible via angiogram in the same location as in the MRI, then an arteriovenous malformation (AVM) becomes the primary concern.
Molecular Mechanisms
Many molecular mechanisms have been identified in CCM pathology. In 2015 it was reported that the endothelial cells forming cerebral vascular malformations undergo an endothelial to mesenchymal transition in both sporadic and familial CCM[12][13]
Incidence
The incidence in the general population is roughly 0.5%, and clinical symptoms typically appear between 20 and 30 years of age.[14] Once thought to be strictly congenital, these vascular lesions have been found to occur de novo. It may appear either sporadically or exhibit autosomal dominant inheritance.
References
- Batra, S.; Lin, D.; Recinos, P. F.; Zhang, J.; Rigamonti, D. (2009). "Cavernous malformations: Natural history, diagnosis and treatment". Nature Reviews Neurology. 5 (12): 659–670. doi:10.1038/nrneurol.2009.177. PMID 19953116. S2CID 11888726.
- Tournier-Lasserve, E.; Jung, S. L. L.; Labauge, H. H.; Houtteville, P.; Lescoat, J. P.; Cecillon, C.; Marechal, M.; Joutel, E.; Bach, A.; Tournier-Lasserve, J. F. O. (1999). "Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas". Nature Genetics. 23 (2): 189–193. doi:10.1038/13815. PMID 10508515. S2CID 22297740.
- Sahoo, T.; Johnson, E. W.; Thomas, J. W.; Kuehl, P. M.; Jones, T. L.; Dokken, C. G.; Touchman, J. W.; Gallione, C. J.; Lee-Lin, S. Q.; Kosofsky, B.; Kurth, J. H.; Louis, D. N.; Mettler, G.; Morrison, L.; Gil-Nagel, A.; Rich, S. S.; Zabramski, J. M.; Boguski, M. S.; Green, E.; Marchuk, D. A. (1999). "Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1)". Human Molecular Genetics. 8 (12): 2325–2333. doi:10.1093/hmg/8.12.2325. PMID 10545614.
- Zawistowski, J. S.; Serebriiskii, I. G.; Lee, M. F.; Golemis, E. A.; Marchuk, D. A. (2002). "KRIT1 association with the integrin-binding protein ICAP-1: A new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis". Human Molecular Genetics. 11 (4): 389–396. doi:10.1093/hmg/11.4.389. PMID 11854171.
- Petersen, T. A.; Morrison, L. A.; Schrader, R. M.; Hart, B. L. (2009). "Familial versus Sporadic Cavernous Malformations: Differences in Developmental Venous Anomaly Association and Lesion Phenotype". American Journal of Neuroradiology. 31 (2): 377–382. doi:10.3174/ajnr.A1822. PMC 4455949. PMID 19833796.
- Günel, M.; Awad, I. A.; Finberg, K.; Anson, J. A.; Steinberg, G. K.; Batjer, H. H.; Kopitnik, T. A.; Morrison, L.; Giannotta, S. L.; Nelson-Williams, C.; Lifton, R. P. (1996). "A Founder Mutation as a Cause of Cerebral Cavernous Malformation in Hispanic Americans". New England Journal of Medicine. 334 (15): 946–951. doi:10.1056/NEJM199604113341503. PMID 8596595.
- Liquori, C. L.; Berg, M. J.; Siegel, A. M.; Huang, E.; Zawistowski, J. S.; Stoffer, T. P.; Verlaan, D.; Balogun, F.; Hughes, L.; Leedom, T. P.; Plummer, N. W.; Cannella, M.; Maglione, V.; Squitieri, F.; Johnson, E. W.; Rouleau, G. A.; Ptacek, L.; Marchuk, D. A. (2003). "Mutations in a Gene Encoding a Novel Protein Containing a Phosphotyrosine-Binding Domain Cause Type 2 Cerebral Cavernous Malformations". The American Journal of Human Genetics. 73 (6): 1459–1464. doi:10.1086/380314. PMC 1180409. PMID 14624391.
- Zawistowski, J. S.; Stalheim, L.; Uhlik, M. T.; Abell, A. N.; Ancrile, B. B.; Johnson, G. L.; Marchuk, D. A. (2005). "CCM1 and CCM2 protein interactions in cell signaling: Implications for cerebral cavernous malformations pathogenesis". Human Molecular Genetics. 14 (17): 2521–2531. doi:10.1093/hmg/ddi256. PMID 16037064.
- Bergametti, F.; Denier, C.; Labauge, P.; Arnoult, M.; Boetto, S.; Clanet, M.; Coubes, P.; Echenne, B.; Ibrahim, R.; Irthum, B.; Jacquet, G.; Lonjon, M.; Moreau, J. J.; Neau, J. P.; Parker, F.; Tremoulet, M.; Tournier-Lasserve, E.; Société Française de Neurochirurgie (2005). "Mutations within the Programmed Cell Death 10 Gene Cause Cerebral Cavernous Malformations". The American Journal of Human Genetics. 76 (1): 42–51. doi:10.1086/426952. PMC 1196432. PMID 15543491.
- Liquori, C. L.; Berg, M. J.; Squitieri, F.; Ottenbacher, M.; Sorlie, M.; Leedom, T. P.; Cannella, M.; Maglione, V.; Ptacek, L.; Johnson, E. W.; Marchuk, D. A. (2006). "Low frequency of PDCD10 mutations in a panel of CCM3 probands: Potential for a fourth CCM locus". Human Mutation. 27 (1): 118. doi:10.1002/humu.9389. PMID 16329096.
- Castro, Marco (2019). "CDC42 Deletion Elicits Cerebral Vascular Malformations via Increased MEKK3-Dependent KLF4 Expression". Circulation Research. 124 (8): 1240–1252. doi:10.1161/CIRCRESAHA.118.314300. PMID 30732528.
- Maddaluno, Luigi (2013). "EndMT contributes to the onset and progression of cerebral cavernous malformations". Nature. 498 (7455): 492–496. Bibcode:2013Natur.498..492M. doi:10.1038/nature12207. PMID 23748444.
- Bravi, Luca (2016). "Endothelial Cells Lining Sporadic Cerebral Cavernous Malformation Cavernomas Undergo Endothelial-to-Mesenchymal Transition". Stroke. 47 (3): 886–890. doi:10.1161/STROKEAHA.115.011867. PMID 26839352.
- Rigamonti, D.; Hadley, M. N.; Drayer, B. P.; Johnson, P. C.; Hoenig-Rigamonti, K.; Knight, J. T.; Spetzler, R. F. (1988). "Cerebral cavernous malformations. Incidence and familial occurrence". The New England Journal of Medicine. 319 (6): 343–347. doi:10.1056/NEJM198808113190605. PMID 3393196.
Further reading
External links
| Classification | |
|---|---|
| External resources |