Catalytic resonance theory
In chemistry, catalytic resonance theory was developed to describe the kinetics of reaction acceleration using dynamic catalyst surfaces. Catalytic reactions occurring on surfaces that undergo variation in surface binding energy and/or entropy exhibit overall increase in reaction rate when the surface binding energy frequencies are comparable to the natural frequencies of the surface reaction, adsorption, and desorption.
History
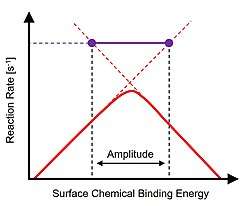
Catalytic resonance theory is constructed on the Sabatier principle of catalysis developed by French chemistry Paul Sabatier. In the limit of maximum catalytic performance, the surface of a catalyst is neither too strong nor too weak. Strong binding results in an overall catalytic reaction rate limitation due to product desorption, while weak binding catalysts are limited in the rate of surface chemistry. Optimal catalyst performance is depicted as a 'volcano' peak using a descriptor of the chemical reaction defining different catalytic materials. Experimental evidence of the Sabatier principle was first demonstrated by Balandin in 1960.[1][2]
The concept of catalytic resonance was proposed on dynamic interpretation of the Sabatier volcano reaction plot.[3] As described, extension of either side of the volcano plot above the peak defines the timescales of the two rate-limiting phenomena such as surface reaction(s) or desorption.[4] For binding energy oscillation amplitudes that extend across the volcano peak, the amplitude endpoints intersect the transiently accessible faster timescales of independent reaction phenomena. At the conditions of sufficiently fast binding energy oscillation, the transient binding energy variation frequency matches the natural frequencies of the reaction and the rate of overall reaction achieves turnover frequencies greatly in excess of the volcano plot peak.[5]
Theory
The basis of catalytic resonance theory utilizes the transient behavior of adsorption, surface reactions, and desorption as surface binding energy and surface transition states oscillate with time. The binding energy of a single species, i, is described via a temporal functional including square or sinusoidal waves of frequency, fi, and amplitude, dUi:

Other surface chemical species, j, are related to the oscillating species, i, by the constant linear parameter, gamma γi-j:

The two surface species also share the common enthalpy of adsorption, delta δi-j. Specification of the oscillation frequency and amplitude of species i and relating γi-j and δi-j for all other surface species j permits determination of all chemical surface species adsorption enthalpy with time. The transition state energy of a surface reaction between any two species i and j is predicted by the linear scaling relationship of the Bell–Evans–Polanyi principle which relates to the surface reaction enthalpy, ΔHi-j, to the transition state energy, Ea, by parameters α and β with the following relationship:

The oscillating surface and transition state energies of chemical species alter the kinetic rate constants associated with surface reaction, adsorption, and desorption. The surface reaction rate constant of species i converting to surface species j includes the dynamic activation energy:

The resulting surface chemistry kinetics are then described via a surface reaction rate expression containing dynamic kinetic parameters responding to the oscillation in surface binding energy:
- ,

with k reactions with dynamic activation energy. The desorption rate constant also varies with oscillating surface binding energy by:
- .

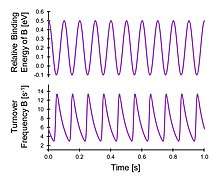
Implementation of dynamic surface binding energy of a reversible A-to-B reaction on a heterogeneous catalyst in a continuous flow stirred tank reactor operating at 1% conversion of A produces a sinusoidal binding energy in species B as shown.[8] In the transition between surface binding energy amplitude endpoints, the instantaneous reaction rate (i.e., turnover frequency) oscillates over an order of magnitude as a limit cycle solution.
Implications for Chemistry
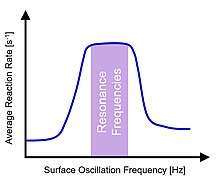
Oscillating binding energies of all surface chemical species introduces periodic instances of transient behavior to the catalytic surface. For slow oscillation frequencies, the transient period is only a small quantity of the oscillation time scale, and the surface reaction achieves a new steady state. However, as the oscillation frequency increases, the surface transient period approaches the timescale of the oscillation and the catalytic surface remains in a constant transient condition. A plot of the averaged turnover frequency of a reaction with respect to applied oscillation frequency identifies the 'resonant' frequency range for which the transient conditions of the catalyst surface match the applied frequencies.[9] The 'resonance band' exists above the Sabatier volcano plot maximum of a static system with average reaction rates as high as five orders of magnitude faster than that achievable by conventional catalysis.
Surface binding energy oscillation also occurs to different extent with the various chemical surface species as defined by the γi-j parameter. For any non-unity γi-j system, the asymmetry in the surface energy profile results in conducting work to bias the reaction to a steady state away from equilibrium.[10] Similar to the controlled directionality of molecular machines, the resulting ratchet (device) energy mechanism selectively moves molecules through a catalytic reaction against a free energy gradient.[11]
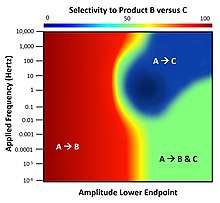
Application of dynamic binding energy to a surface with multiple catalytic reactions exhibits complex behavior derived from the differences in the natural frequencies of each chemistry; these frequencies are identified by the inverse of the adsorption, desorption, and surface kinetic rate parameters. Considering a system of two parallel elementary reactions of A-to-B and A-to-C that only occur on a surface, the performance of the catalyst under dynamic conditions will result in varying capability for selecting either reaction product (B or C).[12] For the depicted system, both reactions have the same overall thermodynamics and will produce B and C in equal amounts (50% selectivity) at chemical equilibrium. Under normal static catalyst operation, only product B can be produced at selectivities greater than 50% and product C is never favored. However, as shown, the application of surface binding dynamics in the form of a square wave at varying frequency and fixed oscillation amplitude but varying endpoints exhibits the full range of possible reactant selectivity. In the range of 1-10 Hertz, there exists a small island of parameters for which product C is highly selective; this condition is only accessible via dynamics. [13]
Experiments and Evidence
In 2020, dynamic oscillation of a catalyst above a static optimum rate was demonstrated for the first time.[14] Using the formic acid electro-oxidation reaction, oscillation of the applied electrodynamic potential between 0 and 0.8 volts accelerated the formation rate of carbon dioxide more than an order of magnitude higher (20X) than what was achievable on platinum, the best existing catalyst. The maximum catalytic rate was experimentally observed at a frequency of 100 Hz; slower catalytic rates were observed at higher and lower electrodynamic frequencies. The resonant frequency was interpreted as the oscillation between conditions favorable to formic acid decomposition (0 V) and conditions favorable to form CO2 (0.8 V).[15]
References
- Helmut Knözinger; Karl Kochloefl (2005). "Heterogeneous Catalysis and Solid Catalysts". Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag. doi:10.1002/14356007.a05_313. ISBN 3527306730.
- Balandin, A. (1969). "Modern State of the Multiplet Theor of Heterogeneous Catalysis1". Adv. Catal. Rel. Subj. Advances in Catalysis. 19: 1–210. doi:10.1016/S0360-0564(08)60029-2. ISBN 9780120078196.
- "Energy Researchers Break the Catalytic Speed Limit". R&D Daily. 2019.
- M.A. Ardagh, O.A. Abdelrahman, P.J. Dauenhauer (2019). "Principles of Dynamic Heterogeneous Catalysis: Surface Resonance and Turnover Frequency Response". ChemRxiv. doi:10.26434/chemrxiv.7790009.v1.CS1 maint: multiple names: authors list (link)
- "Researchers Discover New Technology that can Speed Up Chemical Reactions 10,000 Times Faster". Azo Materials. 2019.
- M.A. Ardagh, O.A. Abdelrahman, P.J. Dauenhauer (2019). "Principles of Dynamic Heterogeneous Catalysis: Surface Resonance and Turnover Frequency Response". ACS Catalysis. 9 (8): 6929–6937. doi:10.1021/acscatal.9b01606.CS1 maint: multiple names: authors list (link)
- M.A. Ardagh, Turan Birol, Q. Zhang, O.A. Abdelrahman, P.J. Dauenhauer (2019). "Catalytic Resonance Theory: superVolcanoes, catalytic molecular pumps, and oscillatory steady state". Catalysis Science & Technology. doi:10.1021/acscatal.9b01606.CS1 maint: multiple names: authors list (link)
- "Breaking the Catalytic Speed Limit". Phys Org. 2019.
- M.A. Ardagh, O.A. Abdelrahman, P.J. Dauenhauer (2019). "Principles of Dynamic Heterogeneous Catalysis: Surface Resonance and Turnover Frequency Response". ACS Catalysis. 9 (8): 6929–6937. doi:10.1021/acscatal.9b01606.CS1 maint: multiple names: authors list (link)
- M.A. Ardagh, Turan Birol, Q. Zhang, O.A. Abdelrahman, P.J. Dauenhauer (2019). "Catalytic Resonance Theory: superVolcanoes, catalytic molecular pumps, and oscillatory steady state". Catalysis Science & Technology. doi:10.1021/acscatal.9b01606.CS1 maint: multiple names: authors list (link)
- Hoffmann, Peter (October 30, 2012). Life's Ratchet: How Molecular Machines Extract Order from Chaos. Basic Books. ISBN 978-0465022533.
- M.A. Ardagh, M. Shetty, A. Kuznetsov, Q. Zhang, P. Christopher, D.G. Vlachos O.A. Abdelrahman, P.J. Dauenhauer (2020). "Catalytic Resonance Theory: Parallel Reaction Pathway Control". Chemical Science. doi:10.1039/C9SC06140A.CS1 maint: multiple names: authors list (link)
- M.A. Ardagh, M. Shetty, A. Kuznetsov, Q. Zhang, P. Christopher, D.G. Vlachos O.A. Abdelrahman, P.J. Dauenhauer (2019). "Catalytic Resonance Theory: Parallel Reaction Pathway Control". ChemRxiv. doi:10.26434/chemrxiv.10271090.v1.CS1 maint: multiple names: authors list (link)
- J. Gopeesingh, M.A. Ardagh, M. Shetty, S.T. Burke, P.J. Dauenhauer, O.A. Abdelrahman (2020). "Resonance-Promoted Formic Acid Oxidation via Dynamic Electrocatalytic Modulation". ACS Catalysis. 9 (8): 6929–6937. doi:10.1021/acscatal.0c02201.CS1 maint: multiple names: authors list (link)
- J. Gopeesingh, M.A. Ardagh, M. Shetty, S.T. Burke, P.J. Dauenhauer, O.A. Abdelrahman (2020). "Resonance-Promoted Formic Acid Oxidation via Dynamic Electrocatalytic Modulation". ChemRxiv. doi:10.26434/chemrxiv.11972031.v1.CS1 maint: multiple names: authors list (link)