CRISPR interference
CRISPR interference (CRISPRi) is a genetic perturbation technique that allows for sequence-specific repression of gene expression in prokaryotic and eukaryotic cells. It was first developed by Stanley Qi and colleagues in the laboratories of Wendell Lim, Adam Arkin, Jonathan Weissman, and Jennifer Doudna.[1] Sequence-specific activation of gene expression refers to CRISPR activation (CRISPRa).
Based on the bacterial genetic immune system - CRISPR (clustered regularly interspaced short palindromic repeats) pathway,[2] the technique provides a complementary approach to RNA interference. The difference between CRISPRi and RNAi, though, is that CRISPRi regulates gene expression primarily on the transcriptional level, while RNAi controls genes on the mRNA level.
Background
Many bacteria and most archaea have an adaptive immune system which incorporates CRISPR RNA (crRNA) and CRISPR-associated (cas) genes.
The CRISPR interference (CRISPRi) technique was first reported by Lei S. Qi and researchers at the University of California at San Francisco in early 2013.[1] The technology uses a catalytically dead Cas9 (usually denoted as dCas9) protein that lacks endonuclease activity to regulate genes in an RNA-guided manner. Targeting specificity is determined by complementary base-pairing of a single guide RNA (sgRNA) to the genomic locus. sgRNA is a chimeric noncoding RNA that can be subdivided into three regions: a 20 nt base-pairing sequence, a 42 nt dCas9-binding hairpin and a 40 nt terminator (bacteria,[3] [4] [5] yeast,[6] fruit flies,[7] zebrafish,[8] mice[9]).
When designing a synthetic sgRNA, only the 20 nt base-pairing sequence is modified. Secondary variables must also be considered: off-target effects (for which a simple BLAST run of the base-pairing sequence is required), maintenance of the dCas9-binding hairpin structure, and ensuring that no restriction sites are present in the modified sgRNA, as this may pose a problem in downstream cloning steps. Due to the simplicity of sgRNA design, this technology is amenable to genome-wide scaling.[10] CRISPRi relies on the generation of catalytically inactive Cas9. This is accomplished by introducing point mutations in the two catalytic residues (D10A and H840A) of the gene encoding Cas9.[11] In doing so, dCas9 is unable to cleave dsDNA but retains the ability to target DNA. Together, sgRNA and dCas9 constitute a minimal system for gene-specific regulation.[1]
Transcriptional regulation
Repression
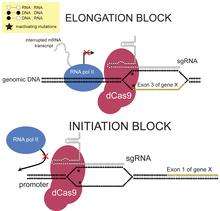
CRISPRi can sterically repress transcription by blocking either transcriptional initiation or elongation. This is accomplished by designing sgRNA complementary to the promoter or the exonic sequences. The level of transcriptional repression for exonic sequences is strand-specific. When targeting the gene body, sgRNA complementary to the non-template strand more strongly represses transcription compared to sgRNA complementary to the template strand. It has been suggested that this is due to the activity of helicase, which unwinds the RNA:DNA heteroduplex ahead of RNA pol II when the sgRNA is complementary to the template strand. Unlike transcription elongation block, silencing is independent of the targeted DNA strand when targeting the transcriptional start site. In prokaryotes, this steric inhibition can repress transcription of the target gene by almost 99.9%; in human cells, up to 90% repression was observed.[1]
CRISPRi can also repress transcription via an effector domain. Fusing a repressor domain to dCas9 allows transcription to be further repressed by inducing heterochromatinization. For example, the well-studied Krüppel associated box (KRAB) domain can be fused to dCas9 to repress transcription of the target gene up to 99% in human cells.[12]
Improvements on the efficiency
Whereas genome-editing by the catalytically active Cas9 nuclease can be accompanied by irreversible off-target genomic alterations, CRISPRi is highly specific with minimal off-target reversible effects for two distinct sgRNA sequences.[12] Nonetheless, several methods have been developed to improve the efficiency of transcriptional modulation. Identification of the transcription start site of a target gene and considering the preferences of sgRNA improves efficiency, as does the presence of accessible chromatin at the target site.[13]
Other methods
Along with other improvements mentioned, factors such as the distance from the transcription start and the local chromatin state may be critical parameters in determining activation/repression efficiency. Optimization of dCas9 and sgRNA expression, stability, nuclear localization, and interaction will likely allow for further improvement of CRISPRi efficiency in mammalian cells.[1]
Applications
Gene knockdown
A significant portion of the genome (both reporter and endogenous genes) in eukaryotes has been shown to be targetable using lentiviral constructs to express dCas9 and sgRNAs, with comparable efficiency to existing techniques such as RNAi and TALE proteins.[12] In tandem or as its own system, CRISPRi could be used to achieve the same applications as in RNAi.
For bacteria, gene knockdown by CRISPRi has been fully implemented and characterized (off-target analysis, leaky repression) for both Gram-negative E. coli [3][5] and Gram-positive B. subtilis.[4]
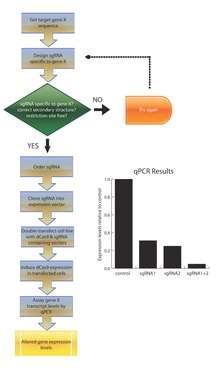
Allelic series
Differential gene expression can be achieved by modifying the efficiency of sgRNA base-pairing to the target loci.[10] In theory, modulating this efficiency can be used to create an allelic series for any given gene, in essence creating a collection of hypo- and hypermorphs. These powerful collections can be used to probe any genetic investigation. For hypomorphs, this allows the incremental reduction of gene function as opposed to the binary nature of gene knockouts and the unpredictability of knockdowns. For hypermorphs, this is in contrast to the conventional method of cloning the gene of interest under promoters with variable strength.
Genome loci imaging
Fusing a fluorescent protein to dCas9 allows for imaging of genomic loci in living human cells.[14] Compared to fluorescence in situ hybridization (FISH), the method uniquely allows for dynamic tracking of chromosome loci. This has been used to study chromatin architecture and nuclear organization dynamics in laboratory cell lines including HeLa cells.
Stem cells
Activation of Yamanaka factors by CRISPRa has been used to induce pluripotency in human and mouse cells providing an alternative method to iPS technology.[15][16] In addition, large-scale activation screens could be used to identify proteins that promote induced pluripotency or, conversely, promote differentiation to a specific cell lineage.[17]
Genetic screening
The ability to upregulate gene expression using dCas9-SunTag with a single sgRNA also opens the door to large-scale genetic screens, such as Perturb-seq, to uncover phenotypes that result from increased or decreased gene expression, which will be especially important for understanding the effects of gene regulation in cancer.[18] Furthermore, CRISPRi systems have been shown to be transferable via horizontal gene transfer mechanisms such as bacterial conjugation and specific repression of reporter genes in recipient cells has been demonstrated. CRISPRi could serve as a tool for genetic screening and potentially bacterial population control.[19]
Advantages and limitations
Advantages
- CRISPRi can silence a target gene of interest up to 99.9% repression.[10]
- Since CRISPRi is based on Watson-Crick base-pairing of sgRNA-DNA and an NGG PAM motif, selection of targetable sites within the genome is straightforward and flexible. Carefully defined protocols have been developed.[10]
- Multiple sgRNAs can not only be used to control multiple different genes simultaneously (multiplex CRISPRi), but also to enhance the efficiency of regulating the same gene target. A popular strategy to express many sgRNAs simultaneously is to array the sgRNAs in a single construct with multiple promoters or processing elements. For example, Extra-Long sgRNA Arrays (ELSAs) use nonrepetitive parts to allow direct synthesis of 12-sgRNA arrays from a gene synthesis provider, can be directly integrated into the E. coli genome without homologous recombination occurring, and can simultaneously target many genes to achieve complex phenotypes.[20]
- While the two systems can be complementary, CRISPRi provides advantages over RNAi. As an exogenous system, CRISPRi does not compete with endogenous machinery such as microRNA expression or function. Furthermore, because CRISPRi acts at the DNA level, one can target transcripts such as noncoding RNAs, microRNAs, antisense transcripts, nuclear-localized RNAs, and polymerase III transcripts. Finally, CRISPRi possesses a much larger targetable sequence space; promoters and, in theory, introns can also be targeted.[12]
- In E. coli, construction of a gene knockdown strain is extremely fast and requires only one-step oligo recombineering.[5]
Limitations
- The requirement of a protospacer adjacent motif (PAM) sequence limits the number of potential target sequences. Cas9 and its homologs may use different PAM sequences, and therefore could theoretically be utilized to expand the number of potential target sequences.[10]
- Sequence specificity to target loci is only 14 nt long (12 nt of sgRNA and 2nt of the PAM), which can recur around 11 times in a human genome.[10] Repression is inversely correlated with the distance of the target site from the transcription start site. Genome-wide computational predictions or selection of Cas9 homologs with a longer PAM may reduce nonspecific targeting.
- Endogenous chromatin states and modifications may prevent the sequence-specific binding of the dCas9-sgRNA complex.[10] The level of transcriptional repression in mammalian cells varies between genes. Much work is needed to understand the role of local DNA conformation and chromatin in relation to binding and regulatory efficiency.
- CRISPRi can influence genes that are in close proximity to the target gene. This is especially important when targeting genes that either overlap other genes (sense or antisense overlapping) or are driven by a bidirectional promoter.[21]
References
- Qi, L. S.; Larson, M. H.; Gilbert, L. A.; Doudna, J. A.; Weissman, J. S.; Arkin, A. P.; Lim, W. A. (2013). "Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression". Cell. 152 (5): 1173–1183. doi:10.1016/j.cell.2013.02.022. PMC 3664290. PMID 23452860.
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D. A.; Horvath, P. (2007). "CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes". Science. 315 (5819): 1709–1712. doi:10.1126/science.1138140. hdl:20.500.11794/38902. PMID 17379808. S2CID 3888761.
- Jiang, W; Bikard, D; Cox, D; Zhang, F; Marraffini, L. A. (2013). "RNA-guided editing of bacterial genomes using CRISPR-Cas systems". Nature Biotechnology. 31 (3): 233–239. doi:10.1038/nbt.2508. PMC 3748948. PMID 23360965.
- Peters, JM; et al. (2016). "A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria". Cell. 165 (6): 1493–1506. doi:10.1016/j.cell.2016.05.003. PMC 4894308. PMID 27238023.
- Li, X; Jun, Y; Erickstad, M; Brown, S; Parks, A; Court, D; Jun, S (2016). "tCRISPRi: tunable and reversible, one-step control of gene expression". Scientific Reports. 6: 39096. doi:10.1038/srep39076. PMC 5171832. PMID 27996021.
- Dicarlo, J. E.; Norville, J. E.; Mali, P; Rios, X; Aach, J; Church, G. M. (2013). "Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems". Nucleic Acids Research. 41 (7): 4336–4343. doi:10.1093/nar/gkt135. PMC 3627607. PMID 23460208.
- Gratz, S. J.; O'Connor-Giles, K. M. (2013). "Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease". Genetics. 194 (4): 1029–1035. doi:10.1534/genetics.113.152710. PMC 3730909. PMID 23709638.
- Hwang, W. Y.; Fu, Y; Reyon, D; Maeder, M. L.; Tsai, S. Q.; Sander, J. D.; Peterson, R. T.; Yeh, J. R.; Joung, J. K. (2013). "Efficient genome editing in zebrafish using a CRISPR-Cas system". Nature Biotechnology. 31 (3): 227–229. doi:10.1038/nbt.2501. PMC 3686313. PMID 23360964.
- Wang, H.; Yang, H.; Shivalila, C. S.; Dawlaty, M. M.; Cheng, A. W.; Zhang, F.; Jaenisch, R. (2013). "One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering". Cell. 153 (4): 910–918. doi:10.1016/j.cell.2013.04.025. PMC 3969854. PMID 23643243.
- Larson, M. H.; Gilbert, L. A.; Wang, X; Lim, W. A.; Weissman, J. S.; Qi, L. S. (2013). "CRISPR interference (CRISPRi) for sequence-specific control of gene expression". Nature Protocols. 8 (11): 2180–2196. doi:10.1038/nprot.2013.132. PMC 3922765. PMID 24136345.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J. A.; Charpentier, E. (2012). "A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity". Science. 337 (6096): 816–821. doi:10.1126/science.1225829. PMC 6286148. PMID 22745249.
- Gilbert, L. A.; Larson, M. H.; Morsut, L; Liu, Z; Brar, G. A.; Torres, S. E.; Stern-Ginossar, N; Brandman, O; Whitehead, E. H.; Doudna, J. A.; Lim, W. A.; Weissman, J. S.; Qi, L. S. (2013). "CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes". Cell. 154 (2): 442–451. doi:10.1016/j.cell.2013.06.044. PMC 3770145. PMID 23849981.
- Radzisheuskaya, Aliaksandra; Shlyueva, Daria; Müller, Iris (28 June 2016). "Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9-mediated transcriptional repression". Nucleic Acids Research. 44 (18): e141. doi:10.1093/nar/gkw583. PMC 5062975. PMID 27353328.
- Chen, B; Gilbert, L. A.; Cimini, B. A.; Schnitzbauer, J; Zhang, W; Li, G. W.; Park, J; Blackburn, E. H.; Weissman, J. S.; Qi, L. S.; Huang, B (2013). "Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system". Cell. 155 (7): 1479–1491. doi:10.1016/j.cell.2013.12.001. PMC 3918502. PMID 24360272.
- Kearns, N. A.; Genga, R. M.; Enuameh, M. S.; Garber, M; Wolfe, S. A.; Maehr, R (2014). "Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells". Development. 141 (1): 219–223. doi:10.1242/dev.103341. PMC 3865759. PMID 24346702.
- Hu, J; Lei, Y; Wong, W. K.; Liu, S; Lee, K. C.; He, X; You, W; Zhou, R; Guo, J. T.; Chen, X; Peng, X; Sun, H; Huang, H; Zhao, H; Feng, B (2014). "Direct activation of human and mouse Oct4 genes using engineered TALE and Cas9 transcription factors". Nucleic Acids Research. 42 (7): 4375–4390. doi:10.1093/nar/gku109. PMC 3985678. PMID 24500196.
- Takahashi, K.; Yamanaka, S. (2006). "Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors". Cell. 126 (4): 663–676. doi:10.1016/j.cell.2006.07.024. hdl:2433/159777. PMID 16904174.
- Tanenbaum, M. E.; Gilbert, L. A.; Qi, L. S.; Weissman, J. S.; Vale, R. D. (2014). "A protein-tagging system for signal amplification in gene expression and fluorescence imaging". Cell. 159 (3): 635–646. doi:10.1016/j.cell.2014.09.039. PMC 4252608. PMID 25307933.
- Ji, W; Lee, D; Wong, E; Dadlani, P; Dinh, D; Huang, V; Kearns, K; Teng, S; Chen, S; Haliburton, J; Heimberg, G; Heineike, B; Ramasubramanian, A; Stevens, T; Helmke, K. J.; Zepeda, V; Qi, L. S.; Lim, W. A. (2014). "Specific gene repression by CRISPRi system transferred through bacterial conjugation". ACS Synthetic Biology. 3 (12): 929–931. doi:10.1021/sb500036q. PMC 4277763. PMID 25409531.
- Reis, Alexander; Halper, Sean; Vezeau, Grace; Cetnar, Daniel; Hossain, Ayaan; Clauer, Phillip; Salis, Howard (2019). "Simultaneous repression of multiple bacterial genes using nonrepetitive extra-long sgRNA arrays". Nature Biotechnology. 37 (11): 1294–1301. doi:10.1038/s41587-019-0286-9. PMID 31591552.
- Goyal, Ashish; Myacheva, Ksenia; Groß, Matthias; Klingenberg, Marcel; Duran Arqué, Berta; Diederichs, Sven (2016-09-30). "Challenges of CRISPR/Cas9 applications for long non-coding RNA genes". Nucleic Acids Research. 45 (3): gkw883. doi:10.1093/nar/gkw883. ISSN 0305-1048. PMC 5388423. PMID 28180319.