Collagen, type III, alpha 1
Type III Collagen is a homotrimer, or a protein composed of three identical peptide chains (monomers), each called an alpha 1 chain of type III collagen. Formally, the monomers are called collagen type III, alpha-1 chain and in humans are encoded by the COL3A1 gene. Type III collagen is one of the fibrillar collagens whose proteins have a long, inflexible, triple-helical domain. [5]
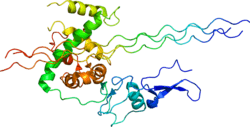




Protein structure and function
Type III collagen is synthesized by cells as a pre-procollagen.[6]
The signal peptide is cleaved off producing a procollagen molecule. Three identical type III procollagen chains come together at the carboxy-terminal ends, and the structure is stabilized by the formation of disulphide bonds. Each individual chain folds into left-handed helix and the three chains are then wrapped together into a right-handed superhelix, the triple helix. Prior to assembling the super-helix, each monomer is subjected to a number of post-translational modifications that occur while the monomer is being translated. First, on the order of 145 prolyl residues of the 239 in the triple-helical domain are hydroxylated to 4-hydroxyproline by prolyl-4-hydroxylase. Second, some of the lysine residues are hydroxylated or glycosylated, and some lysine as well as hydroxylysine residues undergo oxidative deamination catalysed by lysyl oxidase. Other post-translational modifications occur after the triple helix is formed. The large globular domains from both ends of the molecule are removed by C- and amino(N)-terminal-proteinases to generate triple-helical type III collagen monomers called tropocollagen. In addition, crosslinks form between certain lysine and hydroxylysine residues. In the extracellular space in tissues, type III collagen monomers assemble into macromolecular fibrils, which aggregate into fibers, providing a strong support structure for tissues requiring tensile strength.
The triple-helical conformation, which is a characteristic feature of all fibrillar collagens, is possible because of the presence of a glycine as every third amino acid in the sequence of about 1000 amino acids. When the right-handed super-helix is formed, the glycine residues of each of the monomers is positioned at the center of the super-helix (where the three monomers "touch"). Each left-handed helix is characterized by a complete turn in about 3.3 amino acids. The periodicity induced by the glycines at non-integer spacing results in a super-helix that completes one turn in about 20 amino acids. This (Gly-X-Y)n sequence is repeated 343 times in the type III collagen molecule. Proline or hydroxyproline is often found in the X- and Y-position giving the triple helix stability.
In addition to being an integral structural component of many organs, type III collagen is also an important regulator of the diameter of type I and II collagen fibrils. Type III collagen is also known to facilitate platelet aggregation through its binding to platelets and therefore, play an important role in blood clotting.
Tissue distribution
Type III collagen is found as a major structural component in hollow organs such as large blood vessels, uterus and bowel. It is also found in many other tissues together with type I collagen.
Gene
The COL3A1 gene is located on the long (q) arm of chromosome 2 at 2q32.2, between positions 188,974,372 and 189,012,745. The gene has 51 exons and is approximately 40 kbp long.[7] The COL3A1 gene is in tail-to-tail orientation with a gene for another fibrillar collagen, namely COL5A2.[7]
Two transcripts are generated from the gene using different polyadenylation sites. [8] Although alternatively spliced transcripts have been detected for this gene, they are the result of mutations; these mutations alter RNA splicing, often leading to the exclusion of an exon or use of cryptic splice sites. [9][10][11] The resulting defective protein is the cause of a severe, rare disease, the vascular type of Ehlers-Danlos Syndrome (vEDS). These studies have also provided important information about RNA splicing mechanisms in multi-exon genes.[11][9]
Clinical significance
Mutations in the COL3A1 gene cause Ehlers-Danlos syndrome, vascular type (vEDS; also known as the EDS type IV; OMIM 130050). It is the most severe form of EDS, since patients often die suddenly due to rupture of large arteries or other hollow organs.[12]
A few patients with arterial aneurysms without clear signs of EDS have also been found to have COL3A1 mutations.[13][14][15]
More recently, mutations in COL3A1 have also been identified in patients with severe brain anomalies suggesting that type III collagen is important for the normal development of the brain during embryogenesis.[16][17][18][19] This disease is similar to that caused by mutations in GRP56 (OMIM 606854). Type III collagen is a known ligand for the receptor GRP56.
The first single base mutation in the COL3A1 gene was reported in 1989 in a patient with vEDS and changed a glycine amino acid to a serine[20] Since then, over 600 different mutations have been characterized in the COL3A1 gene.[21] About 2/3 of these mutations change a glycine amino acid to another amino acid in the triple-helical region of the protein chain.[12] A large number of RNA splicing mutations have also been identified.[11][9] Interestingly, most of these mutations lead to exon skipping, and produce a shorter polypeptide, in which the Gly-Xaa-Yaa triplets stay in frame and there are no premature termination codons.
The functional consequences of COL3A1 mutations can be studied in a cell culture system. A small bunch biopsy of skin is obtained from the patient and used to start the culture of skin fibroblasts which express type III collagen.[13] The type III collagen protein synthesized by these cells can be studied for its thermal stability. In other words, the collagens can be subjected to a short digestion by proteinases called trypsin and chymotrypsin at increasing temperatures. Intact type III collagen molecules, which have formed a stable triple helix, can withstand such treatment till about 41oC, whereas molecules with mutations that lead to glycine substitutions fall apart at a much lower temperature.
It is difficult to predict the clinical severity based on the type and location of COL3A1 mutations.[22][23] Another important clinical implication is that several studies have reported on mosaicism.[12][24] This refers to a situation where one of the parents carries the mutation in some, but not all of her or his cells, and appears phenotypically healthy, but has more than one affected offspring. In such a situation the risk for another affected child is higher than in a genotypically normal parent.[25]
Type III collagen could also be important in several other human diseases. Increased amounts of type III collagen are found in many fibrotic conditions such as liver and kidney fibrosis, and systemic sclerosis.[26][27][28][29][30][31] This has led to a search for serum biomarkers that could be used for diagnosing these conditions without having to obtain a tissue biopsy. The most widely used biomarker is the N-terminal propeptide of type III procollagen, which is cleaved off during the biosynthesis of type III collagen.[32]
Animal models
Four different mouse models with Col3a1 defects have been reported.[33][34][35][36] Inactivation of the murine Col3a1 gene using homologous recombination technique led to a shorter life span in homozygous mutant mice. The mice died prematurely from a rupture of major arteries mimicking the human vEDS phenotype. These mice also had a severe malformation of the brain. Another study discovered mice with a naturally occurring large deletion of the Col3a1 gene. These mice died suddenly due to thoracic aortic dissections. The third type of mutant mice were transgenic mice with a Gly182Ser mutation. These mice developed severe skin wounds, demonstrated vascular fragility in the form of reduced tensile strength and died prematurely at the age of 13-14 weeks. The fourth mouse model with defective Col3a1 gene is the tight skin mouse (Tsk2/+), which resembles the human systemic sclerosis.
See also
- Collagen
- Ehlers-Danlos syndrome
Notes
References
- GRCh38: Ensembl release 89: ENSG00000168542 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000026043 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Kuivaniemi H, Tromp G (July 2019). "Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases". Gene. 707: 151–171. doi:10.1016/j.gene.2019.05.003. PMID 31075413.
- Kühn K (1989). "Chapter 1: The classical collagens: Types I, II, and III". In Mayne R, Burgeson RE (eds.). Structure and Function of Collagen Types. Orlando, FL: Academic Press. pp. 1–39. ISBN 978-012-481280-2.
- Välkkilä M, Melkoniemi M, Kvist L, Kuivaniemi H, Tromp G, Ala-Kokko L (September 2001). "Genomic organization of the human COL3A1 and COL5A2 genes: COL5A2 has evolved differently than the other minor fibrillar collagen genes". Matrix Biology. 20 (5–6): 357–66. doi:10.1016/s0945-053x(01)00145-7. PMID 11566270.
- Ala-Kokko L, Kontusaari S, Baldwin CT, Kuivaniemi H, Prockop DJ (June 1989). "Structure of cDNA clones coding for the entire prepro alpha 1 (III) chain of human type III procollagen. Differences in protein structure from type I procollagen and conservation of codon preferences". The Biochemical Journal. 260 (2): 509–16. doi:10.1042/bj2600509. PMC 1138697. PMID 2764886.
- Kuivaniemi H, Kontusaari S, Tromp G, Zhao MJ, Sabol C, Prockop DJ (July 1990). "Identical G+1 to A mutations in three different introns of the type III procollagen gene (COL3A1) produce different patterns of RNA splicing in three variants of Ehlers-Danlos syndrome. IV. An explanation for exon skipping some mutations and not others". The Journal of Biological Chemistry. 265 (20): 12067–74. PMID 2365710.
- Kontusaari S, Tromp G, Kuivaniemi H, Ladda RL, Prockop DJ (July 1990). "Inheritance of an RNA splicing mutation (G+ 1 IVS20) in the type III procollagen gene (COL3A1) in a family having aortic aneurysms and easy bruisability: phenotypic overlap between familial arterial aneurysms and Ehlers-Danlos syndrome type IV". American Journal of Human Genetics. 47 (1): 112–20. PMC 1683756. PMID 2349939.
- Schwarze U, Goldstein JA, Byers PH (December 1997). "Splicing defects in the COL3A1 gene: marked preference for 5' (donor) spice-site mutations in patients with exon-skipping mutations and Ehlers-Danlos syndrome type IV". American Journal of Human Genetics. 61 (6): 1276–86. doi:10.1086/301641. PMC 1716081. PMID 9399899.
- Pepin MG, Murray ML, Byers PH (November 2015). Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, Pepin MG, Murray ML, Byers PH (eds.). "Vascular Ehlers-Danlos Syndrome". GeneReviews. PMID 20301667.
- Kontusaari S, Tromp G, Kuivaniemi H, Romanic AM, Prockop DJ (November 1990). "A mutation in the gene for type III procollagen (COL3A1) in a family with aortic aneurysms". The Journal of Clinical Investigation. 86 (5): 1465–73. doi:10.1172/JCI114863. PMC 296891. PMID 2243125.
- Anderson DW, Edwards TK, Ricketts MH, Kuivaniemi H, Tromp G, Stolle CA, Deak SB, Boyd CD (November 1996). "Multiple defects in type III collagen synthesis are associated with the pathogenesis of abdominal aortic aneurysms". Annals of the New York Academy of Sciences. 800: 216–28. doi:10.1111/j.1749-6632.1996.tb33312.x. PMID 8958996.
- Tromp G, Wu Y, Prockop DJ, Madhatheri SL, Kleinert C, Earley JJ, et al. (June 1993). "Sequencing of cDNA from 50 unrelated patients reveals that mutations in the triple-helical domain of type III procollagen are an infrequent cause of aortic aneurysms". The Journal of Clinical Investigation. 91 (6): 2539–45. doi:10.1172/JCI116490. PMC 443315. PMID 8514866.
- Plancke A, Holder-Espinasse M, Rigau V, Manouvrier S, Claustres M, Khau Van Kien P (November 2009). "Homozygosity for a null allele of COL3A1 results in recessive Ehlers-Danlos syndrome". European Journal of Human Genetics. 17 (11): 1411–6. doi:10.1038/ejhg.2009.76. PMC 2986673. PMID 19455184.
- Jørgensen A, Fagerheim T, Rand-Hendriksen S, Lunde PI, Vorren TO, Pepin MG, et al. (June 2015). "Vascular Ehlers-Danlos Syndrome in siblings with biallelic COL3A1 sequence variants and marked clinical variability in the extended family". European Journal of Human Genetics. 23 (6): 796–802. doi:10.1038/ejhg.2014.181. PMC 4795069. PMID 25205403.
- Horn D, Siebert E, Seidel U, Rost I, Mayer K, Abou Jamra R, Mitter D, Kornak U (September 2017). "Biallelic COL3A1 mutations result in a clinical spectrum of specific structural brain anomalies and connective tissue abnormalities". American Journal of Medical Genetics. Part A. 173 (9): 2534–2538. doi:10.1002/ajmg.a.38345. PMID 28742248.
- Vandervore L, Stouffs K, Tanyalçin I, Vanderhasselt T, Roelens F, Holder-Espinasse M, et al. (June 2017). "COL3A1 encoding the ligand to GPR56 are associated with cobblestone-like cortical malformation, white matter changes and cerebellar cysts". Journal of Medical Genetics. 54 (6): 432–440. doi:10.1136/jmedgenet-2016-104421. PMID 28258187.
- Tromp G, Kuivaniemi H, Shikata H, Prockop DJ (January 1989). "A single base mutation that substitutes serine for glycine 790 of the alpha 1 (III) chain of type III procollagen exposes an arginine and causes Ehlers-Danlos syndrome IV". The Journal of Biological Chemistry. 264 (3): 1349–52. PMID 2492273.
- Dalgleish R. "COL3A1". Ehlers Danlos Syndrome Variant Database.
- Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH (December 2014). "Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV)". Genetics in Medicine. 16 (12): 881–8. doi:10.1038/gim.2014.72. PMID 24922459.
- Frank M, Albuisson J, Ranque B, Golmard L, Mazzella JM, Bal-Theoleyre L, Fauret AL, Mirault T, Denarié N, Mousseaux E, Boutouyrie P, Fiessinger JN, Emmerich J, Messas E, Jeunemaitre X (December 2015). "The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome". European Journal of Human Genetics. 23 (12): 1657–64. doi:10.1038/ejhg.2015.32. PMC 4795191. PMID 25758994.
- Richards AJ, Ward PN, Narcisi P, Nicholls AC, Lloyd JC, Pope FM (June 1992). "A single base mutation in the gene for type III collagen (COL3A1) converts glycine 847 to glutamic acid in a family with Ehlers-Danlos syndrome type IV. An unaffected family member is mosaic for the mutation". Human Genetics. 89 (4): 414–8. doi:10.1007/bf00194313. PMID 1352273.
- Kontusaari S, Tromp G, Kuivaniemi H, Stolle C, Pope FM, Prockop DJ (September 1992). "Substitution of aspartate for glycine 1018 in the type III procollagen (COL3A1) gene causes type IV Ehlers-Danlos syndrome: the mutated allele is present in most blood leukocytes of the asymptomatic and mosaic mother". American Journal of Human Genetics. 51 (3): 497–507. PMC 1682722. PMID 1496983.
- Krieg T, Langer I, Gerstmeier H, Keller J, Mensing H, Goerz G, Timpl R (December 1986). "Type III collagen aminopropeptide levels in serum of patients with progressive systemic scleroderma". The Journal of Investigative Dermatology. 87 (6): 788–91. doi:10.1111/1523-1747.ep12459865. PMID 3782862.
- Jimenez SA, Feldman G, Bashey RI, Bienkowski R, Rosenbloom J (August 1986). "Co-ordinate increase in the expression of type I and type III collagen genes in progressive systemic sclerosis fibroblasts". The Biochemical Journal. 237 (3): 837–43. doi:10.1042/bj2370837. PMC 1147064. PMID 3800922.
- Rosenbloom J, Ren S, Macarak E (April 2016). "New frontiers in fibrotic disease therapies: The focus of the Joan and Joel Rosenbloom Center for Fibrotic Diseases at Thomas Jefferson University". Matrix Biology. 51: 14–25. doi:10.1016/j.matbio.2016.01.011. PMID 26807756.
- Fogo AB, Lusco MA, Najafian B, Alpers CE (June 2017). "AJKD Atlas of Renal Pathology: Type III Collagen Glomerulopathy". American Journal of Kidney Diseases. 69 (6): e25–e26. doi:10.1053/j.ajkd.2017.04.004. PMID 28532638.
- Karsdal MA, Nielsen SH, Leeming DJ, Langholm LL, Nielsen MJ, Manon-Jensen T, Siebuhr A, Gudmann NS, Rønnow S, Sand JM, Daniels SJ, Mortensen JH, Schuppan D (November 2017). "The good and the bad collagens of fibrosis - Their role in signaling and organ function". Advanced Drug Delivery Reviews. 121: 43–56. doi:10.1016/j.addr.2017.07.014. PMID 28736303.
- Ricard-Blum S, Baffet G, Théret N (August 2018). "Molecular and tissue alterations of collagens in fibrosis" (PDF). Matrix Biology. 68–69: 122–149. doi:10.1016/j.matbio.2018.02.004. PMID 29458139.
- Risteli J, Niemi S, Trivedi P, Mäentausta O, Mowat AP, Risteli L (April 1988). "Rapid equilibrium radioimmunoassay for the amino-terminal propeptide of human type III procollagen". Clinical Chemistry. 34 (4): 715–8. doi:10.1093/clinchem/34.4.715. PMID 3359606.
- Liu X, Wu H, Byrne M, Krane S, Jaenisch R (March 1997). "Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development". Proceedings of the National Academy of Sciences of the United States of America. 94 (5): 1852–6. doi:10.1073/pnas.94.5.1852. PMC 20006. PMID 9050868.
- Smith LB, Hadoke PW, Dyer E, Denvir MA, Brownstein D, Miller E, et al. (April 2011). "Haploinsufficiency of the murine Col3a1 locus causes aortic dissection: a novel model of the vascular type of Ehlers-Danlos syndrome". Cardiovascular Research. 90 (1): 182–90. doi:10.1093/cvr/cvq356. PMC 3058731. PMID 21071432.
- D'hondt S, Guillemyn B, Syx D, Symoens S, De Rycke R, Vanhoutte L, Toussaint W, Lambrecht BN, De Paepe A, Keene DR, Ishikawa Y, Bächinger HP, Janssens S, Bertrand MJ, Malfait F (September 2018). "Type III collagen affects dermal and vascular collagen fibrillogenesis and tissue integrity in a mutant Col3a1 transgenic mouse model". Matrix Biology. 70: 72–83. doi:10.1016/j.matbio.2018.03.008. PMID 29551664.
- Long KB, Li Z, Burgwin CM, Choe SG, Martyanov V, Sassi-Gaha S, Earl JP, Eutsey RA, Ahmed A, Ehrlich GD, Artlett CM, Whitfield ML, Blankenhorn EP (March 2015). "The Tsk2/+ mouse fibrotic phenotype is due to a gain-of-function mutation in the PIIINP segment of the Col3a1 gene". The Journal of Investigative Dermatology. 135 (3): 718–27. doi:10.1038/jid.2014.455. PMC 4324084. PMID 25330296.
Further reading
- Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. (March 2017). "The 2017 international classification of the Ehlers-Danlos syndromes". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 175 (1): 8–26. doi:10.1002/ajmg.c.31552. PMID 28306229.
- Malfait F (October 2018). "Vascular aspects of the Ehlers-Danlos Syndromes". Matrix Biology. 71–72: 380–395. doi:10.1016/j.matbio.2018.04.013. PMID 29709596.
- Kuivaniemi H, Tromp G, Prockop DJ (November 1991). "Genetic causes of aortic aneurysms. Unlearning at least part of what the textbooks say". The Journal of Clinical Investigation. 88 (5): 1441–4. doi:10.1172/JCI115452. PMC 295644. PMID 1939638.
- Kuivaniemi H, Tromp G, Prockop DJ (1997). "Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels". Human Mutation. 9 (4): 300–15. doi:10.1002/(SICI)1098-1004(1997)9:4<300::AID-HUMU2>3.0.CO;2-9. PMID 9101290.
- Kuivaniemi H, Tromp G, Prockop DJ (April 1991). "Mutations in collagen genes: causes of rare and some common diseases in humans". FASEB Journal. 5 (7): 2052–60. doi:10.1096/fasebj.5.7.2010058. PMID 2010058.
- Byers PH, Belmont J, Black J, De Backer J, Frank M, Jeunemaitre X, Johnson D, Pepin M, Robert L, Sanders L, Wheeldon N (March 2017). "Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 175 (1): 40–47. doi:10.1002/ajmg.c.31553. PMID 28306228.
- Boudko SP, Engel J, Okuyama K, Mizuno K, Bächinger HP, Schumacher MA (November 2008). "Crystal structure of human type III collagen Gly991-Gly1032 cystine knot-containing peptide shows both 7/2 and 10/3 triple helical symmetries". The Journal of Biological Chemistry. 283 (47): 32580–9. doi:10.1074/jbc.M805394200. PMID 18805790.
- Lamberg A, Helaakoski T, Myllyharju J, Peltonen S, Notbohm H, Pihlajaniemi T, Kivirikko KI (May 1996). "Characterization of human type III collagen expressed in a baculovirus system. Production of a protein with a stable triple helix requires coexpression with the two types of recombinant prolyl 4-hydroxylase subunit". The Journal of Biological Chemistry. 271 (20): 11988–95. doi:10.1074/jbc.271.20.11988. PMID 8662631.
External links
- Collagen+type+III at the US National Library of Medicine Medical Subject Headings (MeSH)
- "COL3A1". Ehlers Danlos Syndrome Variant Database.
- "Report for CCDS2297.1". Consensus Coding Sequence (CDS) Database. National Center for Biotechnology Information (NCBI).
- "Ehlers-Danlos Syndrome Type IV". GeneReview. NCBI/NIH/UW.