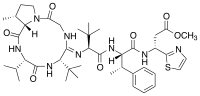
Bottromycin
Bottromycin is a macrocyclic peptide with antibiotic activity. It was first discovered in 1957 as a natural product isolated from Streptomyces bottropensis.[1] It has been shown to inhibit methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococci (VRE) among other Gram-positive bacteria and mycoplasma.[2] Bottromycin is structurally distinct from both vancomycin, a glycopeptide antibiotic, and methicillin, a beta-lactam antibiotic.
 | |
| Names | |
|---|---|
| Other names
Bottromycin A(2); Bottromycic A2 acid, methyl ester | |
| Identifiers | |
3D model (JSmol) |
|
| ChemSpider | |
PubChem CID |
|
| |
| |
| Properties | |
| C42H62N8O7S | |
| Molar mass | 823.05608 g.mol−1 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
Bottromycin binds to the A site of the ribosome and blocks the binding of aminoacyl-tRNA, therefore inhibiting bacterial protein synthesis.[3] Although bottromycin exhibits antibacterial activity in vitro, it has not yet been developed as a clinical antibiotic, potentially due to its poor stability in blood plasma.[2] To increase its stability in vivo, some bottromycin derivatives have been explored.[2]
The structure of bottromycin contains a macrocyclic amidine as well as a thiazole ring. The absolute stereochemistry at several chiral centers has been determined as of 2009.[4] In 2012, a three-dimensional solution structure of bottromycin was published.[5] The solution structure revealed that several methyl groups are on the same face of the structure.
Bottromycin falls within the ribosomally synthesized and post-translationally modified peptide class of natural product.[6]
History
Bottromycin was first isolated from Streptomyces bottropensis in 1957.[1] It has since been identified in at least two other members of the genus Streptomyces;[6][7] members of Streptomyces are known to be prolific producers of secondary metabolites.[8] Bottromycin has a unique structure, consisting of the macrocyclic amidine linkage and four β-methylated amino acids. Bottromycin blocks aminoacyl tRNA binding to the ribosome by binding to the A site of the 50s subunit.[3] Although bottromycin was discovered over 50 years ago, there was a lack of research following initial studies on bottromycin until recent years. The lack of research is potentially a result of bottromycin's low stability in blood plasma.[2] However, the unique structure and mode of action have recently made bottromycin a more target for drug development, especially given the rise of antibiotic resistance.
Mechanism of action
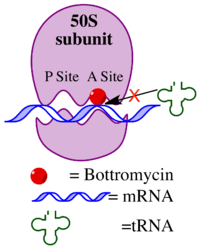
The mechanism of action of bottromycin was confirmed nearly 20 years following the discovery of bottromycin.[3] Bottromycin functions as an antibiotic through inhibition of protein synthesis. It blocks aminoacyl tRNA binding to the ribosome by binding to the A site of the 50s subunit. This results in release of aminoacyl tRNA from the ribosome and premature termination of protein synthesis. A comparison of other antibiotics known to bind to the A site of the ribosome, including micrococcin, tetracycline, streptomycin, and chloramphenicol, suggested that only bottromycin and chloramphenicol caused release of aminoacyl tRNA from the ribosome. Of those antibiotics, only micrococcin is also a macrocyclic peptide.
Structure determination
Bottromycin is produced naturally as a series of products differing in methylation patterns. All products contain valine and phenylalanine methylation. Bottromycin A2 is singly methylated on proline, bottromycin B lacks methylation on proline, and bottromycin C contains a doubly methylated proline.
A partial structure of bottromycin was reported shortly after the initial discovery of bottromycin. The first structural studies relied on traditional methods of analysis. Its peptide-like structure, including the presence of glycine and valine, was first suggested by a combination of acidic hydrolysis, acetylation, ninhydrin staining, and paper chromatography, among other experiments.[9] The presence of a thiazole ring, along with an adjacent β-methylated phenylalanine, was established by ninhydrin staining, potassium permanganate oxidation, and comparison to synthetic standards.[10] A methyl ester substituent was reported in 1958.[11] The same study also reported that the Kunz hydrolysis product lacking a methyl ester was biologically inactive. Nakamura and colleagues later reported that bottromycin contained tert-leucine and cis-3-methylproline.[12] They also proposed a linear iminohexapeptide structure.[13]
These early structural studies were not followed up until recent years with the renewed interest in bottromycin. The structure was confirmed in the 1980s and 1990s to be a cyclic iminopeptide based on NMR studies, with a linear side chain connected to the macrocycle via an amidine linkage.[14][15]
Its absolute stereochemistry, however, was not characterized until 2009.[4] Stereochemistry at carbon 18 and 25 was proposed by comparing predicted conformers obtained using molecular dynamics to experimental constraints obtained through NMR experiments. Stereochemistry at carbon 43 was confirmed by comparing 1H NMR of authentic hydrolysis product to a chemically synthesized sample of the same fragment. Finally, optical rotation, 1H NMR, and HRMS experiments of chemically synthesized bottromycin matched that of biologically produced bottromycin.
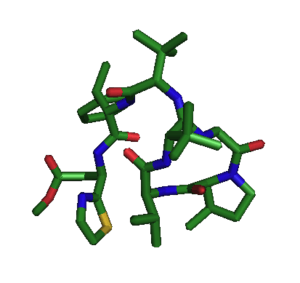
The three-dimensional solution structure of bottromycin A2 was solved by NMR in 2012.[5] The overall structure was obtained with good resolution (RMSD 0.74±0.59 Å), with a RMSD of 0.09±0.06 Å for the macrocycle. In this study, it was proposed that the methylated proline residue contributed to the restricted conformation of the macrocycle. The methylated proline and β-OMe alanine residues were found to be on the same face of bottroymycin A2 and it was suggested that this characteristic contributed to binding of bottromycin to the ribosomal A site.
Biosynthesis
The production of bottromycin by S. bottropensis and S. scabies, as well as the production of a bottromycin analog termed bottromycin D, has been studied.[6][7][16][17] It was independently confirmed in 2012 by multiple groups that bottromycin is produced as a ribosomal peptide natural product that it subsequently post-translationally modified. Before this, it was unclear whether bottromycin was produced by nonribosomal peptide synthetase machinery (NRPS). The presence of amino acids other than the 20 proteinogenic amino acids is often a feature of NRPS products because NRPS machinery can directly incorporate other amino acids, among other chemical building blocks. Ribosomal peptide synthesis, which is the same machinery that produces all proteins found in the cell, is limited to the 20 proteinogenic amino acids. However, bottromycin was found to be a highly modified ribosomal peptide by a combination of genome mining and gene deletion studies.[7][16]
In ribosomal peptide synthesis, the final product results from modifications to a linear peptide starting material translated by the ribosome from an mRNA transcript. In S. scabies the precursor peptide, termed BtmD, is a 44-amino acid peptide.[7] The precursor peptide is termed BmbC in S. bottropensis.[16] The amino acids forming the bottromycin core are residues 2-9 in BtmD: Gly-Pro-Val-Val-Val-Phe-Asp-Cys. In bottromycin D, the sequence is Gly-Pro-Ala-Val-Val-Phe-Asp-Cys, and the precursor peptide is termed BstA.[17] BstA shares high sequence homology with BtmD in the follower peptide region. Unlike other ribosomal peptide natural products, which are normally synthesized with a leader peptide that is cleaved, bottromycin is synthesized with a follower peptide. The presence of a follower peptide was identified by bioinformatic analysis of the bottromycin biosynthetic cluster.
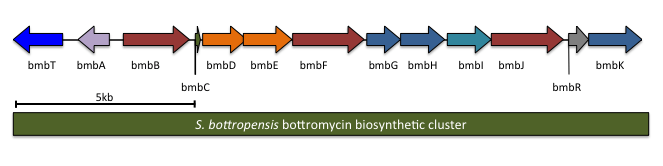
The complete biosynthetic gene cluster for bottromycin has been identified. It is predicted to contain 13 genes, including the precursor peptide (notation will follow Crone and colleagues;[7] other studies had similar results). One of the genes in the cluster, btmL, is proposed to be a transcriptional regulator. Another gene, btmA, is proposed to export bottromycin. The remaining ten genes are expected to modify the precursor peptide btmD from a linear peptide to the final macrocyclic product.
| S. scabies | S. bottropensis | WMMB272 | Predicted function |
|---|---|---|---|
| btmA | bmbT | bstK | Major facilitator superfamily/transporter |
| btmB | bmbA | bstB | o-Methyltransferase |
| btmC | bmbB | bstC | Radical SAM methyltransferase |
| btmD | bmbC | bstA | Precursor peptide |
| btmE | bmbD | bstD | YcaO-domain |
| btmF | bmbE | bstE | YcaO-domain |
| btmG | bmbF | bstF | Radical SAM methyltransferase |
| btmH | bmbG | bstG | α/β Hydrolase |
| btmI | bmbH | bstH | Metallo-dependent hydrolase |
| btmJ | bmbI | bstI | Cytochrome P450 |
| btmK | bmbJ | bstJ | Radical SAM methyltransferase |
| btmL | bmbR | Transcriptional regulator | |
| btmM | bmbK | M17 aminopeptidase |
A biosynthetic pathway has been hypothesized based on proposed gene functions (see figure). btmM, with homology to Zn+2 aminopeptidases, is predicted to cleave the N-terminal methionine residue, which is not present in the bottromycin final product. btmE and btmF both contain YcaO-like domains. It is believed that one Although it is unclear which enzyme is responsible for which step, it is hypothesized that one catalyzes macrocyclic amidine formation while the other catalyzes thiazoline formation. btmJ, encoding an enzyme with cytochrome P450 homology, may oxidize the thiazoline to the thiazole. btmH or btmI both have homology to hydrolytic enzymes (α/β hydrolase and metallo-dependent hydrolase, respectively) may catalyze follower peptide hydrolysis. An alternative proposed role for btmH or btmI is to function as a cyclodehydratase in macrocyclization. Gene deletion studies failed to elucidate the function of other proteins within the cluster.[7]
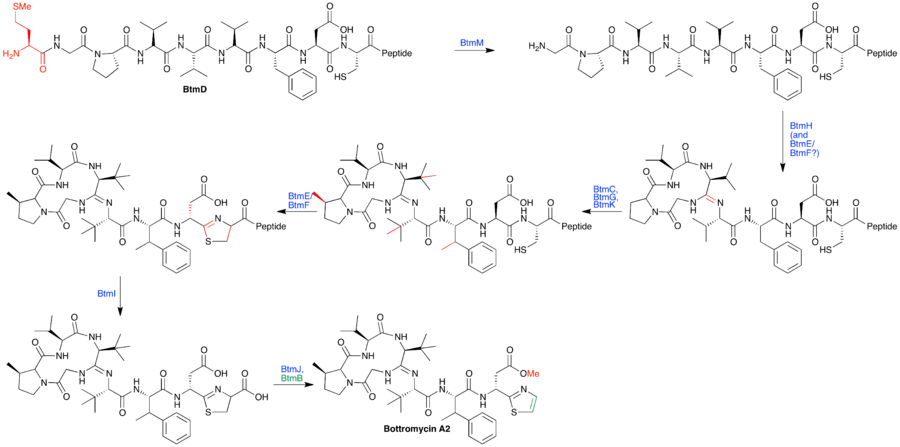
Methyltransferases in the biosynthetic cluster
Bioinformatic analysis identified four methyltransferases within the cluster. Bioinformatics suggest that btmB, is an O-methyltransferase, while the other three, btmC, G and K, are radical S-adenosyl methionine (SAM) methyltransferases. The radical SAM methyltransferases are believed to β-methylate amino acid residues within the precursor peptide. btmC is believed to methylate phenylalanine, btmG is believed to methylate both valines, and btmK is believed to methylate proline based on gene deletion studies.[6][7]
The three putative radical SAM methyltransferases encoded within the pathway are interesting for both mechanistic and biosynthetic reasons. Radical SAM methyltransferases are likely to methylate substrates by an unusual mechanism. Biosynthetically, β-methylations of amino acids are highly unusual in natural products. Polytheonamide B, a peptide natural product produced by a marine symbiont, is the only other structurally characterized example of direct β-methylation of a peptide natural product. The proposed methyl transfer from a SAM-utilizing enzyme was supported by earlier feeding studies with labeled methionine; labeled methionine is used because methionine is converted into SAM within cells.[18] Even further, this study used stereospecifically labeled methionine ([methyl-(2H-3H)]-(2S, methyl-R)-methionine) to show that methylation occurred with a net retention of stereochemistry at the methyl group. The author speculated that net retention indicated a radical mechanism with a B12 intermediate.[18] Radical transfer with a Cobalamin B12 cofactor and SAM has been shown with the few characterized radical SAM methyltransferases. Although the evidence points to radical β-methylation during bottromycin biosynthesis, it remains to be seen whether bioinformatic hypothesis and feeding studies will be supported by in vitro activity assays.
The Val3Ala substitution in bottromycin D does not change the β-methylation pattern between bottromycin A2 and D because Val3 is the only valine not methylated in bottromycin A2. As such, there are still three predicted radical SAM dependent enzymes in the bottromycin D biosynthetic cluster: bstC, bstF, and bstJ.[17]
As of 2013, all published biosynthetic studies have been bioinformatic or cell-based. No biochemical assays directly demonstrating protein function have yet been published. It is likely that in vitro mechanistic studies to better elucidate the biosynthetic pathway will be forthcoming.
Total synthesis
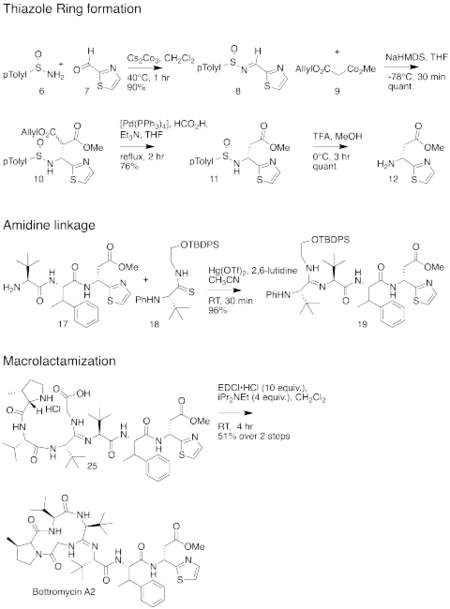
The total synthesis of bottromycin was accomplished in 2009.[4] The synthesis was achieved in 17 steps. Although bottromycin is a peptide-based natural product, it contains an unusual macrocycle and thiazole heterocycle, so that the total synthesis could not be accomplished using traditional solid-phase peptide synthesis. The synthesis was accomplished using a combination of peptide coupling and other methods. To obtain the primary thia-β-Ala-OMe intermediate, a sequence of condensation, Mannich reaction, and palladium-catalyzed decarboxylation steps were performed. This intermediate was prepared stereoselectively. To obtain the amidine linkage, a tripeptide intermediate was coupled to a phthaloyl-protected thioamide via mercury-mediated condensation using mercury (II) trifluoromethanesulfonate (Hg(OTf)2) to yield a branched amidine intermediate. To obtain the final product macrocycle, macrolactamization of the amidine-containing intermediate was required. Macrolactamization was performed with 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and iPr2NEt yielded the final product, bottromycin A2. To confirm that the synthesized bottromycin A2 had the same stereochemistry as natural bottromycin A2, the product was studied by optical rotation, 1H and 13C NMR, IR, and HRMS. The data was found to match that of isolated bottromycin A2. Further, the synthetic sample of bottromycin was also found to have antibacterial activity against both MRSA and VRE, although quantitative data was not reported. A full rendering of the synthetic scheme may be seen under the collapsed synthetic scheme link.
| Bottromycin synthetic scheme as described by Shimamura et al. |
|---|
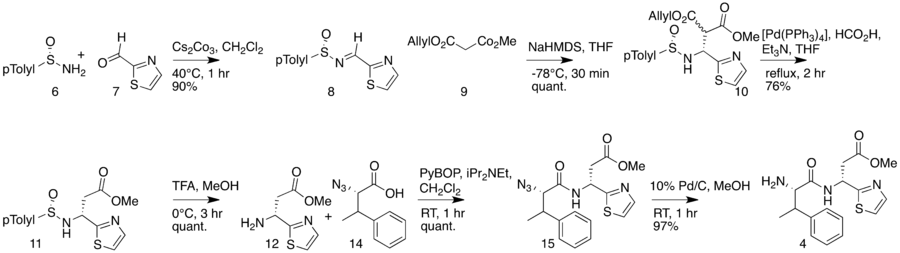 First part of the bottromycin total synthesis as reported by Shimamura et al. 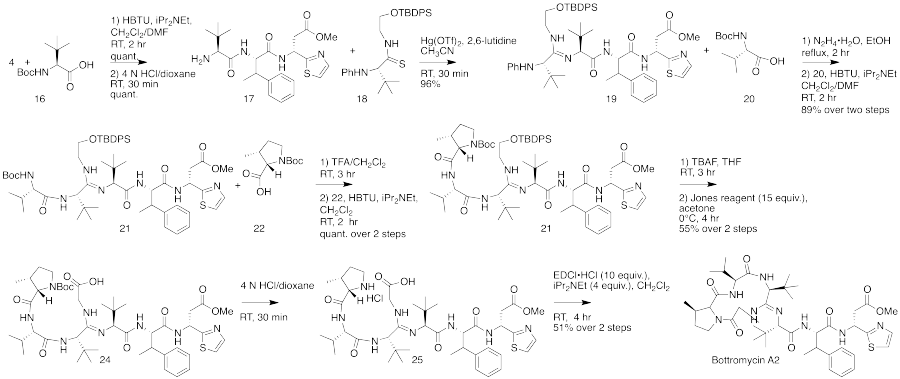 The second part of the bottromycin total synthesis as described by Shimamura et al. |
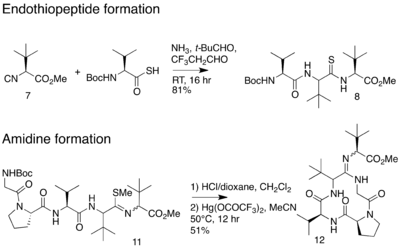
In 2012, an alternative synthesis of the bottromycin macrocyclic ring system and amidine linkage was reported.[19] The synthesis was achieved in 10 steps. Unlike the previous synthesis, Ackerman and colleagues synthesized a linear peptide and achieved intramolecular amidine formation using an S-methylated endothiopeptide. The endothiopeptide was obtained by a thio-Ugi reaction. The resulting macrocycle was obtained as a racemic mixture at the amidine linkage. The full synthetic scheme may be viewed under the collapsed synthetic scheme link.
| Synthetic scheme for alternative bottromycin macrocycle synthesis reported by Ackermann et al. |
|---|
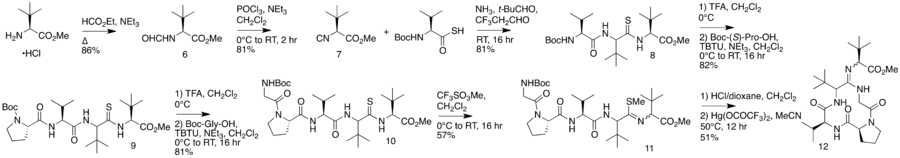 Alternative bottromycin macrocycle synthesis reported by Ackermann et al. |
Derivatives
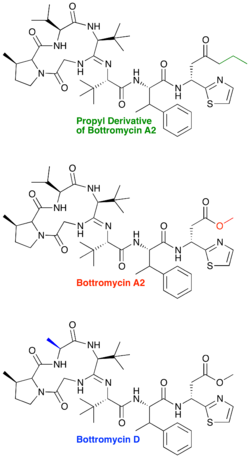
Following the total synthesis of bottromycin, Kobayashi and colleagues synthesized a series of bottromycin derivatives and evaluated their anti-MRSA and anti-VRE activity.[2] Only derivatives of the methyl ester moiety were explored, as they found that the methyl ester was both important for antibacterial activity and unstable in blood plasma. A series of seventeen derivatives were synthesized, with derivatives falling into three general categories: amide derivatives, urea derivatives, and ketone derivatives. All analogs except the carboxylic acid and hydrazide analogs were derivatized from isolated bottromycin A2 using an activated azide ester. The derivatives were tested against six Gram-positive bacterial strains: Staphylococcus aureus FDA209P, S. aureus Smith, MRSA HH-1, MRSA 92-1191, Enterococcus faecalis NCTC12201, and E. faecalis NCTC12203 (both VRE).
Bottromycin A2 had low micromolar activity against all the strains tested, ranging from an MIC of 0.5 μg/mL in E. faecalis NCTC12203 to 2 μg/mL in MRSA HH-1. The amide and urea derivative families were found to have weaker antibacterial activity than bottromycin A2 against S. aureus, MRSA, and VRE. The MIC values for the amide and urea derivatives were generally four times greater than those for bottromycin A2. They were, however, significantly more stable in mouse plasma than bottromycin A2. Bottromycin A2 completely degraded in mouse plasma after 10 minutes and exhibited 0% residual activity after exposure to rat serum. Only one derivative had lower than 50% residual activity. In contrast, many derivatives retained a significant percentage of residual anti-MRSA activity following exposure to serum. Thioester intermediates to the ketone derivatives were found to be unstable, exhibiting 0% residual activity, although they had improved antibacterial activity, exhibiting sub-micromolar MIC values. The propyl ketone was found to be the most promising derivative of all the analogs obtained, both exhibiting antibacterial activity against the bacterial strains tested and stability in plasma, retaining 100% residual activity. The MIC values obtained for the propyl derivative were the same as those found for bottromycin A2 except in the case of NCTC12201, which had an MIC of 2 μg/mL for the derivative and an MIC of 1 μg/mL for bottromycin A2. A summary of MIC values for tested bacterial strains is shown below.
Even the least active bottromycin derivatives exhibited greater anti-VRE activity than vancomycin, which was used as a control antibiotic in this study. The propyl derivative and bottromycin A2 had similar antimicrobial activity to linezolid, a synthetic antibiotic active against Gram-positive bacteria including MRSA and VRE, across all the bacterial strains studied. Overall, the results of this study suggested that further modifications of bottromycin may lead to a more stable, effective antibiotic.
| Compound | S. aureus FDA209P (μg/mL) | S. aureus Smith (μg/mL) | MRSA HH-1 (μg/mL) | MRSA 92-1191 (μg/mL) | VRE NCTC12201 (μg/mL) | VRE NCTC12203 (μg/mL) | Residual anti-MRSA activity (%) |
|---|---|---|---|---|---|---|---|
| Bottromycin A2 | 1 | 1 | 1 | 2 | 1 | 0.5 | 0 |
| Hydrolyzed bottromycin A2 (carboxylic acid) | 64 | 64 | 64 | 128 | 128 | 32 | - |
| Propyl derivative | 1 | 1 | 1 | 2 | 2 | 0.5 | 100 |
| Vancomycin | 1 | 1 | 0.5 | 1 | >128 | >128 | - |
| Linezolid | 2 | 2 | 2 | 2 | 2 | 2 | - |
A natural derivative of bottromycin, bottromycin D, has also been identified.[17] It is produced in a marine Streptomyces species, strain WMMB272. Although the methyl ester is still present in bottromycin D, one of the macrocyclic valines is mutated to an alanine. The minimum inhibitory concentration (MIC) for bottromycin D was determined and found to be only slightly less active than bottromycin A2 (2 μg/mL for bottromycin D vs. 1 μg/mL for bottromycin A2). The authors postulated that greater conformational flexibility of bottromycin D may be responsible for its lower activity.
No further antibacterial studies of synthetic or biosynthetic bottromycin derivatives have been reported in the literature as of 2013. The search for efficacious analogs will be enabled by bottromycin’s status as a ribosomal peptide. Analogs may be explored biosynthetically by changing the sequence of the precursor peptide; a change in amino acid sequence will lead directly to a modified bottromycin structure.
Clinical potential
As of 2013, bottromycin has not been approved for any clinical applications, nor has it been tested in humans. The in vivo stability of bottromycin must be improved before it can be considered as a drug candidate. Work by Kobayashi and colleagues has already begun to address this issue, but more work may be in progress. The need to find new antibiotics to combat antibiotic resistance means that biologic and synthetic interest in bottromycin will likely continue. A combination of biologic and synthetic techniques may yield both an efficacious and stable bottromycin analog for development as a potential drug candidate.
See also
References
- Waisvisz, J. M. (1957). "Bottromycin. I. A New Sulfur-containing Antibiotic". JACS. 79 (16): 4520–4521. doi:10.1021/ja01573a072.
- Kobayashi, Yutaka; et al. (2010). "Bottromycin derivatives: Efficient chemical modifications of the ester moiety and evaluation of anti-MRSA and anti-VRE activities". Bioorganic & Medicinal Chemistry Letters. 20 (20): 6116–6120. doi:10.1016/j.bmcl.2010.08.037. PMID 20833545.
- Otaka, T.; A. Kaji (1976). "Mode of action of bottromycin A2. Release of aminoacyl- or peptidyl-tRNA from ribosomes". Journal of Biological Chemistry. 251 (8): 2299–2306. PMID 770464.
- Shimamura, Hiroyuki; et al. (2009). "Structure Determination and Total Synthesis of Bottromycin A2: A Potent Antibiotic against MRSA and VRE". Angewandte Chemie International Edition. 48 (5): 914–917. doi:10.1002/anie.200804138. PMID 19115340.
- Gouda, Hiroaki; et al. (2012). "Three-dimensional solution structure of bottromycin A2: a potent antibiotic active against methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococci". Chem. Pharm. Bull. 60 (2): 169–171. doi:10.1248/cpb.60.169. PMID 22293474.
- Huo, Liujie; et al. (2012). "Synthetic Biotechnology to Study and Engineer Ribosomal Bottromycin Biosynthesis". Chemistry & Biology. 19 (10): 1278–1287. doi:10.1016/j.chembiol.2012.08.013. PMID 23021914.
- Crone, W. J. K.; F. J. Leeper; A. W. Truman (2012). "Identification and characterisation of the gene cluster for the anti-MRSA antibiotic bottromycin: expanding the biosynthetic diversity of ribosomal peptides". Chemical Science. 3 (12): 3516–3521. doi:10.1039/c2sc21190d.
- Madigan M, Martinko J, eds. (2005). Brock Biology of Microorganisms (11th ed.). Prentice Hall. ISBN 978-0-13-144329-7.
- Waisvisz, J. M.; et al. (1957). "Bottromycin. II. Preliminary degradation studies". JACS. 79 (16): 4522–4524. doi:10.1021/ja01573a073.
- Waisvisz, J. M.; et al. (1957). "The structure of the sulfur-containing moiety of bottromycin". JACS. 79 (16): 4524–4527. doi:10.1021/ja01573a074.
- Waisvisz, J. M.; M. G. Van Der Hoeven (1958). "The Chemistry and Partial Structure of Bottromycin". JACS. 80 (2): 383–385. doi:10.1021/ja01535a034.
- Nakamura, S.; et al. (1965). "Isolation, characterization and structural elucidation of new amino acids from bottromycin A". Chem. Pharm. Bull. 13 (5): 599–602. doi:10.1248/cpb.13.599. PMID 5867718.
- Nakamura, S.; et al. (1966). "Bottromycin A1, A2 and their structures". Journal of Antibiotics. 19 (1): 10–12. PMID 5952015.
- Schipper, D. (1983). "The revised structure of bottromycin A2". Journal of Antibiotics. 36 (8): 1076–1077. doi:10.7164/antibiotics.36.1076. PMID 6630058.
- Kaneda, M. (1992). "Studies on bottromycins. I. 1H and 13C NMR assignments of bottromycin A2, the main component of the complex". Journal of Antibiotics. 45 (5): 792–796. doi:10.7164/antibiotics.45.792. PMID 1624382.
- Gomez-Escribano, Juan; et al. (2012). "Posttranslational β-methylation and macrolactamidination in the biosynthesis of the bottromycin complex of ribosomal peptide antibiotics". Chemical Science. 3 (12): 3522–3525. doi:10.1039/c2sc21183a.
- Hou, Y. (2012). "Structure and Biosynthesis of the Antibiotic Bottromycin D". Organic Letters. 14 (19): 5050–5053. doi:10.1021/ol3022758. PMC 3466018. PMID 22984777.
- Kellenberger, Johannes Laurenz (1997). Zum stereochmischen Verlauf der C-Methylierungsschritte in der Biosynthese von Bottromycin. ETH Zurich: PhD Thesis.
- Ackermann, Stefanie; et al. (2012). "Synthetic studies towards bottromycin". Beilstein Journal of Organic Chemistry. 8: 1652–1656. doi:10.3762/bjoc.8.189. PMC 3510998. PMID 23209498.