Baylis–Hillman reaction
The Baylis–Hillman reaction is a carbon-carbon bond forming reaction between the α-position of an activated alkene and a carbon electrophile such as an aldehyde. Employing a nucleophilic catalyst, such as a tertiary amine and phosphine, this reaction provides a densely functionalized product (e.g. functionalized allyl alcohol in the case of aldehyde as the electrophile).[1][2] It is named for Anthony B. Baylis and Melville E. D. Hillman, two of the chemists who developed this reaction while working at Celanese. This reaction is also known as the Morita–Baylis–Hillman reaction or MBH reaction, as Morita had published earlier work[3] on it.

| (Morita–)Baylis–Hillman reaction | |
|---|---|
| Named after | Ken-ichi Morita Anthony B. Baylis Melville E. D. Hillman |
| Reaction type | Coupling reaction |
| Identifiers | |
| Organic Chemistry Portal | baylis-hillman-reaction |
| RSC ontology ID | RXNO:0000076 |
DABCO is one of the most frequently used tertiary amine catalysts for this reaction. In addition, nucleophilic amines such as DMAP and DBU as well as phosphines have been found to successfully catalyze this reaction.
MBH reaction has several advantages as a useful synthetic method: 1) It is an atom-economic coupling of easily prepared starting materials. 2) Reaction of a pro-chiral electrophile generates a chiral center, therefore an asymmetric synthesis is possible. 3) Reaction products usually contain multiple functionalities in a proximity so that a variety of further transformations are possible. 4) It can employ a nucleophilic organo-catalytic system without the use of heavy metal under mild conditions.
Reaction mechanism
Hoffmann first proposed a mechanism for the MBH reaction.[9] The first reaction step involves 1,4-addition of the catalytic tertiary amine to the activated alkene to generate the zwitterionic aza-enolate. In the second step, this enolate adds to an aldehyde via an aldol addition. The third step involves intramolecular proton shift, which subsequently generates the final MBH adduct and releases the catalyst via E2 or E1cb elimination in the last step. Hill and Isaacs performed kinetic experiments to probe the mechanistic details.[10] The rate of reaction between acrylonitrile and acetaldehyde was first order in concentrations of acrylonitrile, acetaldehyde, and DABCO. Hill and Isaacs proposed that the aldol addition step, which involves all three reactants, thus is the rate determining step. That they did not observe kinetic isotope effect using α-deutrated acrylonitrile also supported this statement.
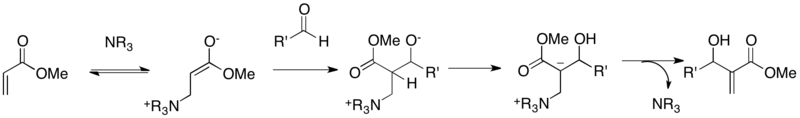
However, this initial mechanistic proposal had been criticized because of several points. The rate of MBH reaction was accelerated by the build-up of product (autocatalytic effect), which could not be rationalized by the mechanism. Also the formation of a considerable amount of ‘unusual’ dioxanone byproduct in the MBH reaction of aryl aldehydes with acrylates was not expected.
McQuade et al. and Aggarwal et al. have reevaluated the MBH mechanism using both kinetics and theoretical studies, focusing on the proton-transfer step.[11][12] According to McQuade, the MBH reaction between methyl acrylate and p-nitrobenzaldehyde is second order relative to the aldehyde and shows a significant kinetic isotope effect at the α-position of the acrylate (5.2 in DMSO). Regardless of the solvents the KIE were found to be greater than 2, indicating the relevance of proton abstraction in the rate-determining step. Based on these new data, McQuade proposed a new mechanism, suggesting that the proton transfer step is the RDS. First and second steps are not changed, but after the first aldol addition the second addition of aldehyde occurs to form a hemiacetal alkoxide. Then the rate-determining proton transfer step via six-membered transition state releases the adduct A, which further reacts to produce MBH product B or dioxanone byproduct C. This mechanism accounts for the formation of dioxanone byproduct.
Aggarwal focused on the autocatalytic effect and observed that the catalytic quantities of MBH product or methanol removed this effect. Thus he proposed that at early stage of the reaction non-alcohol catalyzed mechanism, equivalent to McQuade's proposal, operates, while after 20% conversion alcohol-catalyzed mechanism dominates. In this later stage, alcohol R'OH assists the rate-determining proton transfer step via six membered transition state. Aggarwal and Harvey modeled the two pathways using density functional theory calculations and showed that the computed energy profile matches well with the experimental kinetic isotope effect and observed rate of reaction.[13] Also they showed that the overall enthalpic barrier of the alcohol-catalyzed pathway is slightly smaller than that of the non-alcohol-catalyzed pathway, rationalizing that as the alcohol (MBH product) concentration increases the alcohol-catalyzed pathway starts to dominate, exhibiting the autocatalysis.
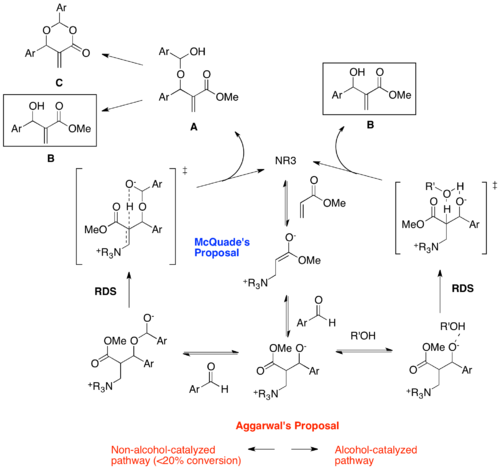
While McQuade's and Aggarwal's studies are receiving much attention recently, there are a number of issues not resolved yet. First, McQuade's proposal for the role of the intermediate A is not clearly proven. Because A could be formed simply by addition of B to an aldehyde, formation of A and C could be happening outside of the MBH mechanism. McQuade asserts that the rate determining step involves two molecules of aldehyde because the reaction rate is second order in aldehyde, but does not explain why Hill and Isaac observed first order for their substrates. Indeed, the enormous variability of substrates for MBH reaction is a constraint for probing the general mechanism of MBH reaction in a unified manner. Also, Aggarwal previously suggested that RDS of the reaction changes from proton transfer to aldol addition over the course of the reaction, based on the fact that primary kinetic isotope effect disappears after 20% conversion,[12] but the subsequent computational studies concluded that the proton transfer step still has the highest barrier in the late stage of reaction. The discrepancy between kinetic and computational results implies that there still are mechanistic aspects of MBH reaction not understood well.
Recently, Coelho and Eberlin et al. have used ESI-MS data to provide experimental data to support the dualistic nature of the reaction's proton transfer step, thus granting the first structural evidence for both McQuade's and Aggarwal's mechanistic propositions for this RDS step of the reaction.[14]
Implications on Asymmetric Catalysis
Nonetheless, the Aggarwal model shed light on the asymmetric catalysis of the MBH reaction. It suggested that all four diastereomers of the intermediate alkoxide are formed in the reaction, but only one has the hydrogen-bond donor suitably positioned to allow fast proton transfer, while the other diastereomers revert to starting materials. These mechanistic studies directed attention to the proton-donor ability (Bronsted acid) of the catalyst. If either the Bronsted acid or the Lewis base could be appropriately positioned on a chiral molecule, the Lewis base would react with the substrate (Michael addition), while the acid in an asymmetric environment would allow the chiral proton transfer. The Bronsted acid remains hydrogen-bonded to the resulting enolate in the enolate-addition step to the aldehyde, and finally ensures efficient proton transfer in the rate-determining proton abstraction step. The action of the Bronsted co-catalysts, which are often employed in MBH reaction, is not limited to a role in proton transfer step. It rather promotes conjugate addition by binding to the zwitterionic enolate, and stabilizing these intermediates.
Scope
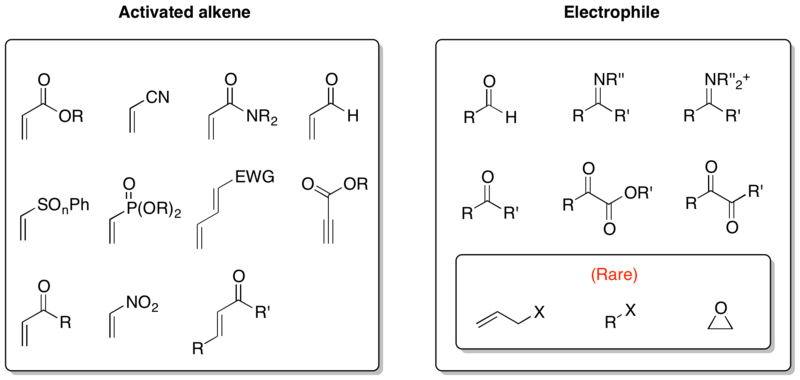
Because the two components of MBH reaction are a general activated alkene and an electrophile, an enormous number of combinations of reaction partners can be generated. Especially, aza-Baylis–Hillman reaction is an important variant of MBH reaction using imines as electrophiles. Although in most cases aldehydes, ketones, or imines are employed as electrophiles, a few reports on the use of allyl halides, alkyl halides, and epoxides have been documented.[15][16][17]
The Baylis–Hillman adducts and their derivatives have been extensively utilized for the generation of heterocycles and other cyclic frameworks.[18]
Using an allene instead of a simple alkene as the precursor for the gives an intermediate that can react at the gamma carbon rather than at the alpha.[19]
Limitations
Because there is a great extent of variability in reaction substrates, it is often challenging to develop reaction conditions suitable for certain combination of substrates. For example, β-substituted activated olefins, vinyl sulfones, and vinyl sulfoxides exhibit low reactivities, slowing or preventing reaction. Competing reactions of substrate functionalities are also problematic. Acroleins are prone to oligomerization, and allenoates easily undergo cycloaddition reactions. It is extremely hard to develop suitable conditions for using alkyl halides and epoxides as electrophiles.
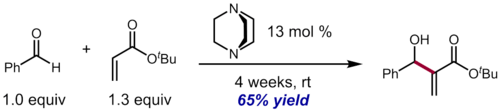
Despite the broad scope, atom economy, and generality of the reaction, the slow rate of the Baylis-Hillman reaction (reaction times of a fortnight or even longer are not uncommon, even with 25 to 100 mol % catalyst) for hindered aliphatic aldehydes and electron-rich benzaldehydes often limits the synthetic utility of the process. For example, in the case of sterically hindered t-butyl acrylate, reaction with benzaldehyde with DABCO as catalyst in the absence of solvent requires 4 weeks to give moderate levels of conversion to the desired product. In the presence of aprotic solvents, the reaction rate is even slower, although protic additives (e.g., alcohols and carboxylic acids) may accelerate the reaction.[20] Ketones are generally not reactive enough to take part in the reaction in a synthetically useful manner under ordinary conditions. However, due to the highly negative volume of activation, sluggish Baylis-Hillman reactions, including those using ketones as substrates, can be realized by conducting the reaction under high pressure (up to 20 kbar).[4]
High reactivity of the activated alkene could also be a problem. The MBH reaction of an aryl vinyl ketone with an aldehyde is not straightforward, since the reactive aryl vinyl ketone readily adds first to another molecule of aryl vinyl ketone via Michael addition, then the adduct adds to the aldehyde to form a double MBH adduct.[21]
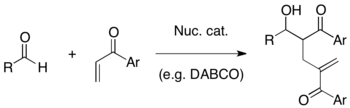
A general solution to asymmetric MBH reaction of diverse substrates is still missing as well. Overall, MBH reaction is not yet at a mature stage, and there is still much room for development of powerful and general catalytic systems.
Variants
Sila-MBH reaction
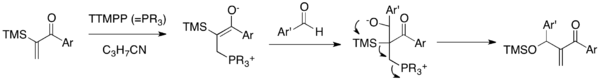
Sila-MBH reaction is a MBH variant that couples α-silylated vinyl aryl ketones with aldehydes in the presence of catalytic TTMPP (Scheme 5).[22] The zwitterionic enolate, produced upon addition of nucleophilic catalyst to enone, would undergo an addition to the carbonyl group of aldehyde to generate an alkoxide. This alkoxide undergoes a subsequent 1,3-Brook rearrangement and elimination cascade to afford a siloxy-methylene enone and release the catalyst. This reaction allows for the synthesis of syloxy-methylene aryl enones, the class which was unavailable via a traditional MBH reaction. Importantly, this reaction overcomes the double MBH addition problem of aryl vinyl ketones.
Rauhut-Currier reaction
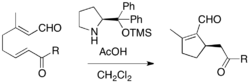
Rauhut-Currier reaction is a reaction of activated alkene and a Michael acceptor, not an aldehyde or an imine. It is also called a vinylogous MBH reaction. Because Rauhut-Currier reaction often couple two activated alkenes, there have been problems with selectivity. Intramolecular Rauhut-Currier reaction has been employed by the virtue of improved reactivity and selectivity. For example, Rauhut-Currier cyclization of α,β-unsaturated aldehydes can be performed in the presence of proline derivative and acetic acid, affording enantioenriched products.[23]
Tandem reaction / Multicomponent one-pot reaction
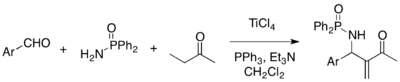
Multicomponent reaction strategy is attractive for its atom-economical virtue. MBH reaction can be employed for three-component coupling of aldehydes, amines, and activated alkenes to afford aza-MBH adducts. For example, reactions of aryl aldehydes, diphenylphosphinamide, and methyl vinyl ketone, in the presence of TiCl4, triphenylphosphine, and triethylamine, give the corresponding aza-MBH adducts.[24]
In addition, activated acetylenes can be added to electrophiles following a Michael addition. Trimethylsilyl iodide as the Michael donor can perform three-component reaction, while tandem cyclization is also possible via Michael attack of a moiety in the MBH electrophile.[25]
Asymmetric MBH reaction
Chiral Auxiliary
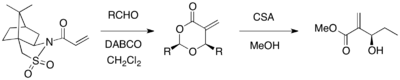
Oppolzer's sultam can be used as a chiral auxiliary for an asymmetric MBH reaction. When an acrylate substituted with the Oppolzer's sultam reacted with various aldehydes in the presence of DABCO catalyst, optically pure 1,3-dioxan-4-ones were afforded with cleavage of the auxiliary (67-98% yield, >99% ee). The cyclic products could be converted into desired MBH products by use of CSA and methanol.[26]
A related hydrazide auxiliary can also be used for similar DABCO-catalyzed MBH reaction. The chiral acryloylhydrazide can react with aldehydes diastereoselectively.[27] Both diastereomers could be obtained from the same reactants by the different choice of solvents (DMSO yielded one diastereomer, while THF/H2O yielded the other one), suggesting that the transition structure conformation is solvent-dependent.
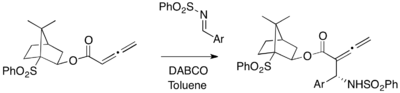
Chiral allenes and imines can be employed for an asymmetric DABCO-catalyzed aza-MBH reaction.[28] Optically active 10-phenylsulfonylisobornyl buta-2,3-dienoate reacts with aryl imine to afford α-allenylamine in a diastereoselective manner (37-57% yield).
Chiral Lewis base catalyst
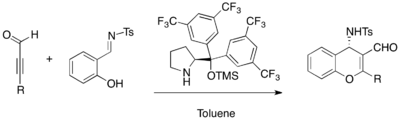
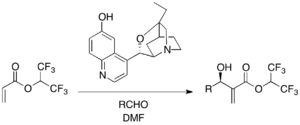
Chiral tertiary amine catalysts are employed for enantioselective MBH reactions. β-ICD, a cinchona alkaloid derivative, is famous among the quinidine framework-based catalysts. 1,1,1,3,3,3,-hexafluoroisopropyl acrylate as an activated alkene and various aldehydes undergo MBH reaction in the presence of β-ICD.[29] The phenolic oxygen of β-ICD was shown to be important in the reaction, implying the function of Bronsted acid moiety. β-ICD and its related versions are effective catalysts for various other substrates.
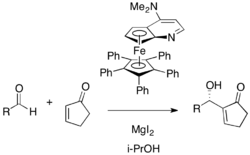
Cyclopentenone and various aromatic and aliphatic aldehydes undergo an asymmetric reaction using Fu's planar chiral DMAP catalyst in isopropanol (54-96% yield, 53-98% ee). In this case, magnesium iodide as a Lewis acid cocatalyst was required to accelerate the reaction.[30] P-Chiral phosphines have been investigated.[31]
Simple diamines can also be employed as MBH catalysts. Methyl vinyl ketone and various substituted benzaldehydes were found to undergo asymmetric MBH reaction. The chiral pyrrolidine catalyst was effective for ortho- and para-substituted electron-deficient benzaldehydes (75-99% yield, 8-73% ee).[32]
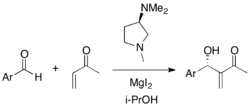
Chiral phosphine MBH catalysts often contain Bronsted acid moiety in their backbones. For example, chiral phosphines containing a Lewis base, a Bronsted acid, and an acid-activated Bronsted base were developed for an asymmetric aza-MBH reaction (86-96% yield, 79-92% ee). The Bronsted acid and base moieties were proposed to be involved in the stabilization of zwitterionic species in a stereoselective manner.[33]
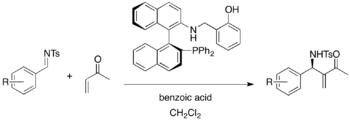
BINOL-derived chiral phosphine catalyst is also effective for an asymmetric aza-MBH reaction of N-tosyl imines with activated alkenes such as methyl vinyl ketone and phenyl acrylate.[34]
In addition, a distinct class of chiral phosphine-squaramide molecules could effectively catalyze an intramolecular asymmetric MBH reaction. ω-formylenones reacted to afford enantioenriched cyclic products at ambient temperature (64-98% yield, 88-93% ee).[35]
Chiral Lewis acid catalyst
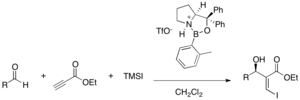
Chiral Lewis acid catalysts have been given interests as they could activate the electron-withdrawing group in an enantioselective manner. Chiral cationic oxazaborolidinium catalysts were shown to be effective in the three-component coupling of α,β-acetylenic esters, aldehydes, and trimethylsilyl iodide (50-99% yield, 62-94% ee). Both enantiomeric products could be obtained by using different enantiomers of the catalyst.[36]
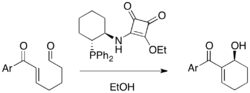
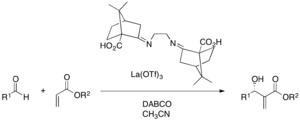
Complex of metal salt and chiral ligand is a viable strategy, too. La(OTf)3 and camphor-derived chiral ligands could induce enantioselectivity in a DABCO-catalyzed MBH reaction of various aldehydes and acrylates (25-97% yield, 6-95% ee). For these cases, multidentate ligands were usually employed to chelate with the metal, which activates both the zwitterionic enolate and the aldehyde.[37]
La(O-iPr)3 and BINOL-derived ligand system, in conjunction with catalytic DABCO, also works for an asymmetric aza-MBH reaction of various N-diphenylphosphinoyl imines and methyl acrylate. Aryl, heteroaryl, and alkenyl imines were all suitable for good yield and enantioselectivity.[38]
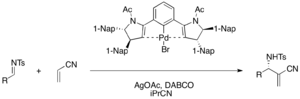
Chiral palladium(II) pincer complexes function as Lewis acid in the enantioselective DABCO-catalyzed aza-MBH reaction of acrylonitrile and various tosyl imines to afford functionalized α-methylene-β-aminonitriles (75-98% yield, 76-98% ee). Silver acetate is required to activate the palladium bromide precatalyst in the catalytic cycle.[39]
Chiral Bronsted acid cocatalyst
A variety of chiral thiourea catalysts are under investigation for asymmetric MBH reactions. Chiral thiourea and bis(thiourea) catalysts can be effective in DABCO-catalyzed MBH and aza-MBH reactions.[40][41] Jacobsen's thiourea catalyst performs an enantioselective aza-MBH reaction, for example (25-49% yield, 87-99% ee).
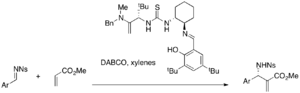
While simple thiourea requires a nucleophilic catalyst in conjunction, bifunctional catalysts such as phosphine-thioureas can be used alone for asymmetric MBH reactions. For example, various acrylates and aromatic aldehydes react in the presence of these catalysts to afford either enantiomeric MBH adducts (32-96% yield, 9-77% ee).[42]
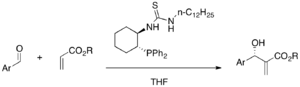
MBH reaction can involve proline derivative as a cocatalyst. It was proposed that imidazole nucleophilic catalyst and proline effect the reaction via iminium intermediate.[43] With (S)-proline and DABCO, α-amido sulfones and α,β-unsaturated aldehydes undergo a highly enantioselective aza-MBH reaction (46-87% yield, E/Z 10:1-19:1, 82-99% ee).[44]
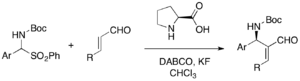
Applications in Organic Synthesis
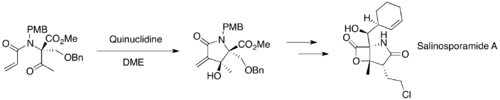
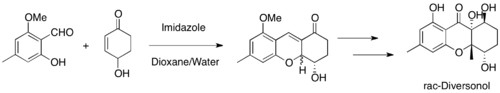
MBH reactions are widely used in organic synthesis. For example, this reaction was used to construct key cyclic intermediates for syntheses of salinosporamide A, diversonol, and anatoxin-a.[45][46][47]
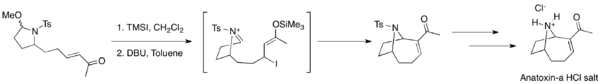
References
- Baylis, A. B.; Hillman, M. E. D. German Patent 2155113, 1972.
- Ciganek, E. Org. React. 1997, 51, 201. doi:10.1002/0471264180.or051.02
- K. Morita, Z. Suzuki and H. Hirose, Bull. Chem. Soc. Jpn.,1968, 41, 2815.
- Recent Advances in the Baylis−Hillman Reaction and Applications Deevi Basavaiah, Anumolu Jaganmohan Rao, and Tummanapalli Satyanarayana Chem. Rev., 2003, 103 (3), pp 811–892 2003 (Article) doi:10.1021/cr010043d
- Masson, G., Housseman, C. and Zhu, J. (2007), The Enantioselective Morita–Baylis–Hillman Reaction and Its Aza Counterpart. Angewandte Chemie International Edition, 46: 4614–4628. doi:10.1002/anie.200604366
- aza-Baylis−Hillman Reaction Valerie Declerck, Jean Martinez and Frederic Lamaty Chem. Rev., 2009, 109 (1), pp 1–48, 2009 (Review) doi:10.1021/cr068057c
- Recent Contributions from the Baylis−Hillman Reaction to Organic Chemistry Deevi Basavaiah, Bhavanam Sekhara Reddy and Satpal Singh Badsara Chemical Reviews 2010 110 (9), 5447-5674 doi:10.1021/cr900291g
- The Baylis–Hillman reaction: a novel concept for creativity in chemistry Deevi Basavaiah and Gorre Veeraraghavaiah Chem. Soc. Rev., 2012, Advance Article doi:10.1039/C1CS15174F
- Angew. Chem. Int. Ed. Engl. 1983, 22, 795.
- J. Phys. Org. Chem. 1990, 3, 285.
- Organic Letters, 2005, 7, 1, 147-150.
- Angew. Chem. Int. Ed. 2005, 44, 1706-1708.
- J. Am. Chem. Soc. 2007, 129, 15513.
- J. Org. Chem., 2009, 74(8), 3031-3037
- Tetrahedron Lett. 2001, 42, 85.
- Org. Lett. 2010, 12, 2418.
- Chem. Commun. 2006, 2977.
- Tetrahedron, 2008, 64(20), 4511-4574.
- J. Am. Chem. Soc. 2009, 131, 4196.
- Fort, Yves; Berthe, Marie Christine; Caubere, Paul (1992). "The 'Baylis - Hillman Reaction' mechanism and applications revisited". Tetrahedron. 48 (31): 6371–6384. doi:10.1016/s0040-4020(01)88227-2.
- Angew. Chem. Int. Ed. 2012, 51, 10337.
- Organic Letters, 2009, 11, 1, 253-255.
- Org. Lett. 2009, 11, 4116.
- Tetrahedron Lett., 2002, 43, 9171.
- Chem. Eur. J. 2010, 16, 9453
- J. Am. Chem. Soc. 1997, 119, 4317-4318
- Org. Lett. 2000, 2, 6, 729-731
- Eur. J. Org. Chem. 2010, 3249-3256
- J. Am. Chem. Soc. 1999, 121, 10219-10220
- Chem. Commun. 2010, 46, 2644-2646
- Xiao, Y.; Sun, Z.; Guo, H.; Kwon, O. (2014). "Chiral Phosphines in Nucleophilic Organocatalysis". Beilstein Journal of Organic Chemistry. 10: 2089–2121. doi:10.3762/bjoc.10.218. PMC 4168899. PMID 25246969.CS1 maint: uses authors parameter (link)
- J. Tetrahedron: Asymmetry, 2010, 1511.
- Adv. Synth. Catal. 2009, 351, 331
- Chem. Commun. 2003, 1310
- Chem. Commun. 2011, 47, 1012
- Angew. Chem. Int. Ed. 2009, 48, 4398
- J. Org. Chem. 2003, 68, 915-919
- J. Am. Chem. Soc. 2010, 132, 11988
- Angew. Chem. Int. Ed. 2012, 51, 10337-10341
- Adv. Synth. Catal. 2005, 347, 1701-1708
- Tetrahedron Lett. 2011, 52, 6234
- Tetrahedron 2009, 65, 8185
- Chem. Eur, J. 2009, 15, 1734
- J. Adv. Synth. Catal. 2011, 353, 1096
- J. Am. Chem. Soc. 2004, 126, 6230-6231.
- Angew. Chem. Int. Ed. 2006, 45, 307–309.
- Chem. Commun. 2008, 3432.