Bacterial genome
Bacterial genomes are generally smaller and less variant in size among species when compared with genomes of eukaryotes. Bacterial genomes can range in size anywhere from about 130 kbp[1][2] to over 14 Mbp.[3] A study that included, but was not limited to, 478 bacterial genomes, concluded that as genome size increases, the number of genes increases at a disproportionately slower rate in eukaryotes than in non-eukaryotes. Thus, the proportion of non-coding DNA goes up with genome size more quickly in non-bacteria than in bacteria. This is consistent with the fact that most eukaryotic nuclear DNA is non-gene coding, while the majority of prokaryotic, viral, and organellar genes are coding.[4] Right now, we have genome sequences from 50 different bacterial phyla and 11 different archaeal phyla. Second-generation sequencing has yielded many draft genomes (close to 90% of bacterial genomes in GenBank are currently not complete); third-generation sequencing might eventually yield a complete genome in a few hours. The genome sequences reveal much diversity in bacteria. Analysis of over 2000 Escherichia coli genomes reveals an E. coli core genome of about 3100 gene families and a total of about 89,000 different gene families.[5] Genome sequences show that parasitic bacteria have 500–1200 genes, free-living bacteria have 1500–7500 genes, and archaea have 1500–2700 genes.[6] A striking discovery by Cole et al. described massive amounts of gene decay when comparing Leprosy bacillus to ancestral bacteria.[7] Studies have since shown that several bacteria have smaller genome sizes than their ancestors did.[8] Over the years, researchers have proposed several theories to explain the general trend of bacterial genome decay and the relatively small size of bacterial genomes. Compelling evidence indicates that the apparent degradation of bacterial genomes is owed to a deletional bias.
Methods and techniques
As of 2014, there are over 30,000 sequenced bacterial genomes publicly available and thousands of metagenome projects. Projects such as the Genomic Encyclopedia of Bacteria and Archaea (GEBA) intend to add more genomes.[5]
The single gene comparison is now being supplanted by more general methods. These methods have resulted in novel perspectives on genetic relationships that previously have only been estimated.[5]
A significant achievement in the second decade of bacterial genome sequencing was the production of metagenomic data, which covers all DNA present in a sample. Previously, there were only two metagenomic projects published.[5]
Bacterial genomes
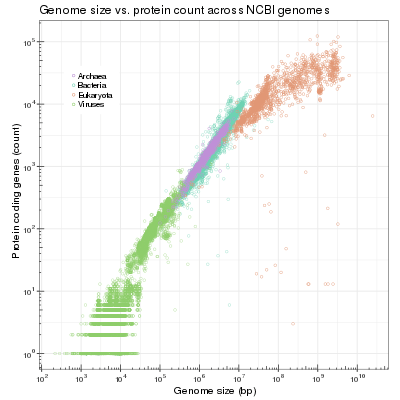
Bacteria possess a compact genome architecture distinct from eukaryotes in two important ways: bacteria show a strong correlation between genome size and number of functional genes in a genome, and those genes are structured into operons.[9][10] The main reason for the relative density of bacterial genomes compared to eukaryotic genomes (especially multicellular eukaryotes) is the presence of noncoding DNA in the form of intergenic regions and introns.[10] Some notable exceptions include recently formed pathogenic bacteria. This was initially described in a study by Cole et al. in which Mycobacterium leprae was discovered to have a significantly higher percentage of pseudogenes to functional genes (~40%) than its free-living ancestors.[7]
Furthermore, amongst species of bacteria, there is relatively little variation in genome size when compared with the genome sizes of other major groups of life.[6] Genome size is of little relevance when considering the number of functional genes in eukaryotic species. In bacteria, however, the strong correlation between the number of genes and the genome size makes the size of bacterial genomes an interesting topic for research and discussion.[11]
The general trends of bacterial evolution indicate that bacteria started as free-living organisms. Evolutionary paths led some bacteria to become pathogens and symbionts. The lifestyles of bacteria play an integral role in their respective genome sizes. Free-living bacteria have the largest genomes out of the three types of bacteria; however, they have fewer pseudogenes than bacteria that have recently acquired pathogenicity.
Facultative and recently evolved pathogenic bacteria exhibit a smaller genome size than free-living bacteria, yet they have more pseudogenes than any other form of bacteria.
Obligate bacterial symbionts or pathogens have the smallest genomes and the fewest pseudogenes of the three groups.[12] The relationship between life-styles of bacteria and genome size raises questions as to the mechanisms of bacterial genome evolution. Researchers have developed several theories to explain the patterns of genome size evolution amongst bacteria.
Genome comparisons and phylogeny
As single-gene comparisons have largely given way to genome comparisons, phylogeny of bacterial genomes have improved in accuracy. The Average Nucleotide Identity method quantifies genetic distance between entire genomes by taking advantage of regions of about 10,000 bp. With enough data from genomes of one genus, algorithms are executed to categorize species. This has been done for the Pseudomonas avellanae species in 2013.[5]
To extract information about bacterial genomes, core- and pan-genome sizes have been assessed for several strains of bacteria. In 2012, the number of core gene families was about 3000. However, by 2015, with an over tenfold increased in available genomes, the pan-genome has increased as well. There is roughly a positive correlation between the number of genomes added and the growth of the pan-genome. On the other hand, the core genome has remain static since 2012. Currently, the E. coli pan-genome is composed of about 90,000 gene families. About one-third of these exist only in a single genome. Many of these, however, are merely gene fragments and the result of calling errors. Still, there are probably over 60,000 unique gene families in E. coli.[5]
Theories of bacterial genome evolution
Bacteria lose a large amount of genes as they transition from free-living or facultatively parasitic life cycles to permanent host-dependent life. Towards the lower end of the scale of bacterial genome size are the mycoplasmas and related bacteria. Early molecular phylogenetic studies revealed that mycoplasmas represented an evolutionary derived state, contrary to prior hypotheses. Furthermore, it is now known that mycoplasmas are just one instance of many of genome shrinkage in obligately host-associated bacteria. Other examples are Rickettsia, Buchnera aphidicola, and Borrelia burgdorferi.[13]
Small genome size in such species is associated with certain particularities, such as rapid evolution of polypeptide sequences and low GC content in the genome. The convergent evolution of these qualities in unrelated bacteria suggests that an obligate association with a host promotes genome reduction.[13]
Given that over 80% of almost all of the fully sequenced bacterial genomes consist of intact ORFs, and that gene length is nearly constant at ~1 kb per gene, it is inferred that small genomes have few metabolic capabilities. While free-living bacteria, such as E. coli, Salmonella species, or Bacillus species, usually have 1500 to 6000 proteins encoded in their DNA, obligately pathogenic bacteria often have as few as 500 to 1000 such proteins.[13]
One candidate explanation is that reduced genomes maintain genes that are necessary for vital processes pertaining to cellular growth and replication, in addition to those genes that are required to survive in the bacteria's ecological niche. However, sequence data contradicts this hypothesis. The set of universal orthologs amongst eubacteria comprises only 15% of each genome. Thus, each lineage has taken a different evolutionary path to reduced size. Because universal cellular processes require over 80 genes, variation in genes imply that the same functions can be achieved by exploitation of nonhomologous genes.[13]
Host-dependent bacteria are able to secure many compounds required for metabolism from the host's cytoplasm or tissue. They can, in turn, discard their own biosynthetic pathways and associated genes. This removal explains many of the specific gene losses. For example, the Rickettsia species, which relies on specific energy substrate from its host, has lost many of its native energy metabolism genes. Similarly, most small genomes have lost their amino acid biosynthesizing genes, as these are found in the host instead. One exception is the Buchnera, an obligate maternally transmitted symbiont of aphids. It retains 54 genes for biosynthesis of crucial amino acids, but no longer has pathways for those amino acids that the host can synthesize. Pathways for nucleotide biosynthesis are gone from many reduced genomes. Those anabolic pathways that evolved through niche adaptation remain in particular genomes.[13]
The hypothesis that unused genes are eventually removed does not explain why many of the removed genes would indeed remain helpful in obligate pathogens. For example, many eliminated genes code for products that are involved in universal cellular processes, including replication, transcription, and translation. Even genes supporting DNA recombination and repair are deleted from every small genome. In addition, small genomes have fewer tRNAs, utilizing one for several amino acids. So, a single codon pairs with multiple codons, which likely yields less-than-optimal translation machinery. It is unknown why obligate intracellular pathogens would benefit by retaining fewer tRNAs and fewer DNA repair enzymes.[13]
Another factor to consider is the change in population that corresponds to an evolution towards an obligately pathogenic life. Such a shift in lifestyle often results in a reduction in the genetic population size of a lineage, since there is a finite number of hosts to occupy. This genetic drift may result in fixation of mutations that inactivate otherwise beneficial genes, or otherwise may decrease the efficiency of gene products. Hence, not will only useless genes be lost (as mutations disrupt them once the bacteria has settled into host dependency), but also beneficial genes may be lost if genetic drift enforces ineffective purifying selection.[13]
The number of universally maintained genes is small and inadequate for independent cellular growth and replication, so that small genome species must achieve such feats by means of varying genes. This is done partly through nonorthologous gene displacement. That is, the role of one gene is replaced by another gene that achieves the same function. Redundancy within the ancestral, larger genome is eliminated. The descendant small genome content depends on the content of chromosomal deletions that occur in the early stages of genome reduction.[13]
The very small genome of M. genitalium possesses dispensable genes. In a study in which single genes of this organism were inactivated using transposon-mediated mutagenesis, at least 129 of its 484 ORGs were not required for growth. A much smaller genome than that of the M. genitalium is therefore feasible.[13]
Doubling time
One theory predicts that bacteria have smaller genomes due to a selective pressure on genome size to ensure faster replication. The theory is based upon the logical premise that smaller bacterial genomes will take less time to replicate. Subsequently, smaller genomes will be selected preferentially due to enhanced fitness. A study done by Mira et al. indicated little to no correlation between genome size and doubling time.[14] The data indicates that selection is not a suitable explanation for the small sizes of bacterial genomes. Still, many researchers believe there is some selective pressure on bacteria to maintain small genome size.
Deletional bias
Selection is but one process involved in evolution. Two other major processes (mutation and genetic drift) can account for the genome sizes of various types of bacteria. A study done by Mira et al. examined the size of insertions and deletions in bacterial pseudogenes. Results indicated that mutational deletions tend to be larger than insertions in bacteria in the absence of gene transfer or gene duplication.[14] Insertions caused by horizontal or lateral gene transfer and gene duplication tend to involve transfer of large amounts of genetic material. Assuming a lack of these processes, genomes will tend to reduce in size in the absence of selective constraint. Evidence of a deletional bias is present in the respective genome sizes of free-living bacteria, facultative and recently derived parasites and obligate parasites and symbionts.
Free-living bacteria tend to have large population-sizes and are subject to more opportunity for gene transfer. As such, selection can effectively operate on free-living bacteria to remove deleterious sequences resulting in a relatively small number of pseudogenes. Continually, further selective pressure is evident as free-living bacteria must produce all gene-products independent of a host. Given that there is sufficient opportunity for gene transfer to occur and there are selective pressures against even slightly deleterious deletions, it is intuitive that free-living bacteria should have the largest bacterial genomes of all bacteria types.
Recently-formed parasites undergo severe bottlenecks and can rely on host environments to provide gene products. As such, in recently-formed and facultative parasites, there is an accumulation of pseudogenes and transposable elements due to a lack of selective pressure against deletions. The population bottlenecks reduce gene transfer and as such, deletional bias ensures the reduction of genome size in parasitic bacteria.
Obligatory parasites and symbionts have the smallest genome sizes due to prolonged effects of deletional bias. Parasites which have evolved to occupy specific niches are not exposed to much selective pressure. As such, genetic drift dominates the evolution of niche-specific bacteria. Extended exposure to deletional bias ensures the removal of most superfluous sequences. Symbionts occur in drastically lower numbers and undergo the most severe bottlenecks of any bacterial type. There is almost no opportunity for gene transfer for endosymbiotic bacteria, and thus genome compaction can be extreme. One of the smallest bacterial genomes ever to be sequenced is that of the endosymbiont Carsonella rudii.[15] At 160 kbp, the genome of Carsonella is one of the most streamlined examples of a genome examined to date.
Genomic reduction
Molecular phylogenetics has revealed that every clade of bacteria with genome sizes under 2 Mb was derived from ancestors with much larger genomes, thus refuting the hypothesis that bacteria evolved by the successive doubling of small-genomed ancestors.[16] Recent studies performed by Nilsson et al. examined the rates of bacterial genome reduction of obligate bacteria. Bacteria were cultured introducing frequent bottlenecks and growing cells in serial passage to reduce gene transfer so as to mimic conditions of endosymbiotic bacteria. The data predicted that bacteria exhibiting a one-day generation time lose as many as 1,000 kbp in as few as 50,000 years (a relatively short evolutionary time period). Furthermore, after deleting genes essential to the methyl-directed DNA mismatch repair (MMR) system, it was shown that bacterial genome size reduction increased in rate by as much as 50 times.[17] These results indicate that genome size reduction can occur relatively rapidly, and loss of certain genes can speed up the process of bacterial genome compaction.
This is not to suggest that all bacterial genomes are reducing in size and complexity. While many types of bacteria have reduced in genome size from an ancestral state, there are still a huge number of bacteria that maintained or increased genome size over ancestral states.[8] Free-living bacteria experience huge population sizes, fast generation times and a relatively high potential for gene transfer. While deletional bias tends to remove unnecessary sequences, selection can operate significantly amongst free-living bacteria resulting in evolution of new genes and processes.
Horizontal gene transfer
Unlike eukaryotes, which evolve mainly through the modification of existing genetic information, bacteria have acquired a large percentage of their genetic diversity by the horizontal transfer of genes. This creates quite dynamic genomes, in which DNA can be introduced into and removed from the chromosome.[18]
Bacteria have more variation in their metabolic properties, cellular structures, and lifestyles than can be accounted for by point mutations alone. For example, none of the phenotypic traits that distinguish E. coli from Salmonella enterica can be attributed to point mutation. On the contrary, evidence suggests that horizontal gene transfer has bolstered the diversification and speciation of many bacteria.[18]
Horizontal gene transfer is often detected via DNA sequence information. DNA segments obtained by this mechanism often reveal a narrow phylogenetic distribution between related species. Furthermore, these regions sometimes display an unexpected level of similarity to genes from taxa that are assumed to be quite divergent.[18]
Although gene comparisons and phylogenetic studies are helpful in investigating horizontal gene transfer, the DNA sequences of genes are even more revelatory of their origin and ancestry within a genome. Bacterial species differ widely in overall GC content, although the genes in any one species' genome are roughly identical with respect to base composition, patterns of codon usage, and frequencies of di- and trinucleotides. As a result, sequences that are newly acquired through lateral transfer can be identified via their characteristics, which remains that of the donor. For example, many of the S. enterica genes that are not present in E. coli have base compositions that differ from the overall 52% GC content of the entire chromosome. Within this species, some lineages have more than a megabase of DNA that is not present in other lineages. The base compositions of these lineage-specific sequences imply that at least half of these sequences were captured through lateral transfer. Furthermore, the regions adjacent to horizontally obtained genes often have remnants of translocatable elements, transfer origins of plasmids, or known attachment sites of phage integrases.[18]
In some species, a large proportion of laterally transferred genes originate from plasmid-, phage-, or transposon-related sequences.[18]
Although sequence-based methods reveal the prevalence of horizontal gene transfer in bacteria, the results tend to be underestimates of the magnitude of this mechanism, since sequences obtained from donors whose sequence characteristics are similar to those of the recipient will avoid detection.[18]
Comparisons of completely sequenced genomes confirm that bacterial chromosomes are amalgams of ancestral and laterally acquired sequences. The hyperthermophilic Eubacteria Aquifex aeolicus and Thermotoga maritima each has many genes that are similar in protein sequence to homologues in thermophilic Archaea. 24% of Thermotoga's 1,877 ORFs and 16% of Aquifex's 1,512 ORFs show high matches to an Archaeal protein, while mesophiles such as E. coli and B. subtilis have far lesser proportions of genes that are most like Archaeal homologues.[18]
Mechanisms of lateral transfer
The genesis of new abilities due to horizontal gene transfer has three requirements. First, there must exist a possible route for the donor DNA to be accepted by the recipient cell. Additionally, the obtained sequence must be integrated with the rest of the genome. Finally, these integrated genes must benefit the recipient bacterial organism. The first two steps can be achieved via three mechanisms: transformation, transduction and conjugation.[18]
Transformation involves the uptake of named DNA from the environment. Through transformation, DNA can be transmitted between distantly related organisms. Some bacterial species, such as Haemophilus influenzae and Neisseria gonorrhoeae, are continuously competent to accept DNA. Other species, such as Bacillus subtilis and Streptococcus pneumoniae, become competent when they enter a particular phase in their lifecycle.
Transformation in N. gonorrhoeae and H. influenzae is effective only if particular recognition sequences are found in the recipient genomes (5'-GCCGTCTGAA-3' and 5'-AAGTGCGGT-3'. respectively). Although the existence of certain uptake sequences improve transformation capability between related species, many of the inherently competent bacterial species, such as B. subtilis and S. pneumoniae, do not display sequence preference.
New genes may be introduced into bacteria by a bacteriophage that has replicated within a donor through generalized transduction or specialized transduction. The amount of DNA that can be transmitted in one event is constrained by the size of the phage capsid (although the upper limit is about 100 kilobases). While phages are numerous in the environment, the range of microorganisms that can be transduced depends on receptor recognition by the bacteriophage. Transduction does not require both donor and recipient cells to be present simultaneously in time nor space. Phage-encoded proteins both mediate the transfer of DNA into the recipient cytoplasm and assist integration of DNA into the chromosome.[18]
Conjugation involves physical contact between donor and recipient cells and is able to mediate transfers of genes between domains, such as between bacteria and yeast. DNA is transmitted from donor to recipient either by self-transmissible or mobilizable plasmid. Conjugation may mediate the transfer of chromosomal sequences by plasmids that integrate into the chromosome.
Despite the multitude of mechanisms mediating gene transfer among bacteria, the process's success is not guaranteed unless the received sequence is stably maintained in the recipient. DNA integration can be sustained through one of many processes. One is persistence as an episome, another is homologous recombination, and still another is illegitimate incorporation through lucky double-strand break repair.[18]
Traits introduced through lateral gene transfer
Antimicrobial resistance genes grant an organism the ability to grow its ecological niche, since it can now survive in the presence of previously lethal compounds. As the benefit to a bacterium earned from receiving such genes are time- and space-independent, those sequences that are highly mobile are selected for. Plasmids are quite mobilizable between taxa and are the most frequent way by which bacteria acquire antibiotic resistance genes.
Adoption of a pathogenic lifestyle often yields a fundamental shift in an organism's ecological niche. The erratic phylogenetic distribution of pathogenic organisms implies that bacterial virulence is a consequence of the presence, or obtainment of, genes that are missing in avirulent forms. Evidence of this includes the discovery of large 'virulence' plasmids in pathogenic Shigella and Yersinia, as well as the ability to bestow pathogenic properties onto E. coli via experimental exposure to genes from other species.[18]
Computer-made form
In April 2019, scientists at ETH Zurich reported the creation of the world's first bacterial genome, named Caulobacter ethensis-2.0, made entirely by a computer, although a related viable form of C. ethensis-2.0 does not yet exist.[19][20]
See also
References
- McCutcheon, J. P.; Von Dohlen, C. D. (2011). "An Interdependent Metabolic Patchwork in the Nested Symbiosis of Mealybugs". Current Biology. 21 (16): 1366–1372. doi:10.1016/j.cub.2011.06.051. PMC 3169327. PMID 21835622.
- Van Leuven, JT; Meister, RC; Simon, C; McCutcheon, JP (11 September 2014). "Sympatric speciation in a bacterial endosymbiont results in two genomes with the functionality of one". Cell. 158 (6): 1270–80. doi:10.1016/j.cell.2014.07.047. PMID 25175626.
- Han, K; Li, ZF; Peng, R; Zhu, LP; Zhou, T; Wang, LG; Li, SG; Zhang, XB; Hu, W; Wu, ZH; Qin, N; Li, YZ (2013). "Extraordinary expansion of a Sorangium cellulosum genome from an alkaline milieu". Scientific Reports. 3: 2101. Bibcode:2013NatSR...3E2101H. doi:10.1038/srep02101. PMC 3696898. PMID 23812535.
- Hou, Yubo; Lin, Senjie (2009). "Distinct Gene Number-Genome Size Relationships for Eukaryotes and Non-Eukaryotes: Gene Content Estimation for Dinoflagellate Genomes". PLOS ONE. 4 (9): e6978. Bibcode:2009PLoSO...4.6978H. doi:10.1371/journal.pone.0006978. PMC 2737104. PMID 19750009.
- Land, Miriam; Hauser, Loren; Jun, Se-Ran; Nookaew, Intawat; Leuze, Michael R.; Ahn, Tae-Hyuk; Karpinets, Tatiana; Lund, Ole; Kora, Guruprased; Wassenaar, Trudy; Poudel, Suresh; Ussery, David W. (2015). "Insights from 20 years of bacterial genome sequencing". Functional & Integrative Genomics. 15 (2): 141–161. doi:10.1007/s10142-015-0433-4. PMC 4361730. PMID 25722247.

- Gregory, T. R. (2005). "Synergy between sequence and size in Large-scale genomics". Nature Reviews Genetics. 6 (9): 699–708. doi:10.1038/nrg1674. PMID 16151375.
- Cole, S. T.; Eiglmeier, K.; Parkhill, J.; James, K. D.; Thomson, N. R.; Wheeler, P. R.; Honoré, N.; Garnier, T.; Churcher, C.; Harris, D.; Mungall, K.; Basham, D.; Brown, D.; Chillingworth, T.; Connor, R.; Davies, R. M.; Devlin, K.; Duthoy, S.; Feltwell, T.; Fraser, A.; Hamlin, N.; Holroyd, S.; Hornsby, T.; Jagels, K.; Lacroix, C.; MacLean, J.; Moule, S.; Murphy, L.; Oliver, K.; Quail, M. A. (2001). "Massive gene decay in the leprosy bacillus". Nature. 409 (6823): 1007–1011. Bibcode:2001Natur.409.1007C. doi:10.1038/35059006. PMID 11234002.
- Ochman, H. (2005). "Genomes on the shrink". Proceedings of the National Academy of Sciences. 102 (34): 11959–11960. Bibcode:2005PNAS..10211959O. doi:10.1073/pnas.0505863102. PMC 1189353. PMID 16105941.
- Gregory, T. Ryan (2005). The evolution of the genome. Burlington, MA: Elsevier Academic. ISBN 0123014638.
- Koonin, E. V. (2009). "Evolution of genome architecture". The International Journal of Biochemistry & Cell Biology. 41 (2): 298–306. doi:10.1016/j.biocel.2008.09.015. PMC 3272702. PMID 18929678.
- Kuo, C. -H.; Moran, N. A.; Ochman, H. (2009). "The consequences of genetic drift for bacterial genome complexity". Genome Research. 19 (8): 1450–1454. doi:10.1101/gr.091785.109. PMC 2720180. PMID 19502381.
- Ochman, H.; Davalos, L. M. (2006). "The Nature and Dynamics of Bacterial Genomes". Science. 311 (5768): 1730–1733. Bibcode:2006Sci...311.1730O. doi:10.1126/science.1119966. PMID 16556833.
- Moran, Nancy A. (2002). "Microbial Minimalism". Cell. 108 (5): 583–586. doi:10.1016/S0092-8674(02)00665-7. PMID 11893328.
- Mira, A.; Ochman, H.; Moran, N. A. (2001). "Deletional bias and the evolution of bacterial genomes". Trends in Genetics. 17 (10): 589–596. doi:10.1016/S0168-9525(01)02447-7. PMID 11585665.
- Nakabachi, A.; Yamashita, A.; Toh, H.; Ishikawa, H.; Dunbar, H. E.; Moran, N. A.; Hattori, M. (2006). "The 160-Kilobase Genome of the Bacterial Endosymbiont Carsonella". Science. 314 (5797): 267. doi:10.1126/science.1134196. PMID 17038615.
- Ochman, H. (2005). "Genomes on the shrink". Proceedings of the National Academy of Sciences. 102 (34): 11959–11960. Bibcode:2005PNAS..10211959O. doi:10.1073/pnas.0505863102. PMC 1189353. PMID 16105941.
- Nilsson, A. I.; Koskiniemi, S.; Eriksson, S.; Kugelberg, E.; Hinton, J. C.; Andersson, D. I. (2005). "Bacterial genome size reduction by experimental evolution". Proceedings of the National Academy of Sciences. 102 (34): 12112–12116. Bibcode:2005PNAS..10212112N. doi:10.1073/pnas.0503654102. PMC 1189319. PMID 16099836.
- Ochman, Howard; Lawrence, Jeffrey G.; Groisman, Eduardo A. (2000). "Lateral gene transfer and the nature of bacterial innovation". Nature. 405 (6784): 299–304. Bibcode:2000Natur.405..299O. doi:10.1038/35012500. PMID 10830951.
- ETH Zurich (1 April 2019). "First bacterial genome created entirely with a computer". EurekAlert!. Retrieved 2 April 2019.
- Venetz, Jonathan E.; et al. (1 April 2019). "Chemical synthesis rewriting of a bacterial genome to achieve design flexibility and biological functionality". Proceedings of the National Academy of Sciences of the United States of America. 116 (16): 8070–8079. doi:10.1073/pnas.1818259116. PMC 6475421. PMID 30936302.