Sertoli–Leydig cell tumour
Sertoli–Leydig cell tumour is a group of tumors composed of variable proportions of Sertoli cells, Leydig cells, and in the case of intermediate and poorly differentiated neoplasms, primitive gonadal stroma and sometimes heterologous elements.[1]
| Sertoli–Leydig cell tumour | |
|---|---|
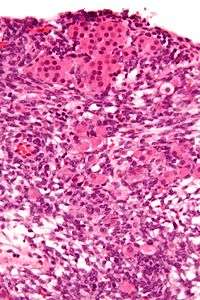 | |
| Micrograph of a Sertoli–Leydig cell tumour. The Leydig cells have abundant eosinophilic or light pink cytoplasm. The Sertoli cells have a pale/clear cytoplasm. H&E stain. | |
| Specialty | Endocrinology, oncology |
Sertoli–Leydig cell tumour (a sex-cord stromal tumor), is a testosterone-secreting ovarian tumor and is a member of the sex cord-stromal tumour group[2] of ovarian and testicular cancers. The tumour occurs in early adulthood (not seen in newborn), is rare, comprising less than 1% of testicular tumours.[1] While the tumour can occur at any age, it occurs most often in young adults. Recent studies have shown that many cases of Sertoli–Leydig cell tumor of the ovary are caused by germline mutations in the DICER1 gene.[3][4] These hereditary cases tend to be younger, often have a multinodular thyroid goiter and there may be a personal or family history of other rare tumors such as pleuropulmonary blastoma, Wilms tumor and cervical rhabdomyosarcoma.
Closely related terms include arrhenoblastoma[5] and androblastoma.[6] Both terms are classified under Sertoli–Leydig cell tumour in MeSH.
Signs and symptoms
Due to excess testosterone secreted by the tumour, one-third of adult females present with a recent history of progressive masculinization. Masculinization is preceded by anovulation, oligomenorrhoea, amenorrhoea and defeminization. Additional signs include acne and hirsutism, voice deepening, clitoromegaly, temporal hair recession, and an increase in musculature. Serum testosterone level is high.
Cause
The exact cause of Sertoli–Leydig cell tumour is not known. Research studies seem to indicate that certain genetic mutations (in the DICER1 gene) may play a role in many cases.
Diagnosis
Presence of an ovarian tumour plus hormonal disturbances suggests a Sertoli–Leydig cell tumour. However, hormonal disturbance is present in only two-thirds of cases. A conclusive diagnosis is made via histology, as part of a pathology report made during or after surgery. See also sex cord–gonadal stromal tumour.
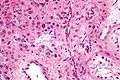 High magnification micrograph of a Leydig cell tumour. H&E stain.
High magnification micrograph of a Leydig cell tumour. H&E stain.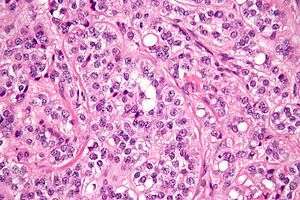
Classification
The tumour is subdivided into many different subtypes. The most typical is composed of tubules lined by Sertoli cells and interstitial clusters of Leydig cells.
Treatment
The usual treatment is surgery. The surgery usually is a fertility-sparing unilateral salpingo-oophorectomy. For malignant tumours, the surgery may be radical and usually is followed by adjuvant chemotherapy, sometimes by radiation therapy. In all cases, initial treatment is followed by surveillance. Because in many cases Sertoli–Leydig cell tumour does not produce elevated tumour markers,[7] the focus of surveillance is on repeated physical examination and imaging. Given that many cases of Sertoli–Leydig cell tumor of the ovary are hereditary, referral to a clinical genetics service should be considered.
The prognosis is generally good as the tumour tends to grow slowly and usually is benign: 25% are malignant. For malignant tumours with undifferentiated histology, prognosis is poor.[7]
See also
- Androgen-dependent syndromes
- Leydig cell tumour
- Sertoli cell tumour
References
- WHO, 2003
- Sachdeva, Poonam; Arora, Raksha; Dubey, Chandan; Sukhija, Astha; Daga, Mridula; Kumar Singh, Deepak (2008). "Sertoli–Leydig cell tumor: A rare ovarian neoplasm. Case report and review of literature". Gynecological Endocrinology. 24 (4): 230–4. doi:10.1080/09513590801953465. PMID 18382911.
- Frio, Thomas Rio; Bahubeshi, Amin; Kanellopoulou, Chryssa; Hamel, Nancy; Niedziela, Marek; Sabbaghian, Nelly; Pouchet, Carly; Gilbert, Lucy; O'Brien, Paul K. (2011). "DICER1 Mutations in Familial Multinodular Goiter With and Without Ovarian Sertoli-Leydig Cell Tumors". JAMA. 305 (1): 68–77. doi:10.1001/jama.2010.1910. PMC 3406486. PMID 21205968.
- Slade, Ingrid; Bacchelli, Chiara; Davies, Helen; Murray, Anne; Abbaszadeh, Fatemeh; Hanks, Sandra; Barfoot, Rita; Burke, Amos; Chisholm, Julia (2011). "DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome". Journal of Medical Genetics. 48 (4): 273–8. doi:10.1136/jmg.2010.083790. PMID 21266384.
- "arrhenoblastoma" at Dorland's Medical Dictionary
- "androblastoma" at Dorland's Medical Dictionary
- Lenhard, Miriam; Kuemper, Caroline; Ditsch, Nina; Diebold, Joachim; Stieber, Petra; Friese, Klaus; Burges, Alexander (2007). "Use of novel serum markers in clinical follow-up of Sertoli–Leydig cell tumours". Clinical Chemistry and Laboratory Medicine. 45 (5): 657–61. doi:10.1515/CCLM.2007.120. PMID 17484630.
External links

| Classification | |
|---|---|
| External resources |
}}