Amyloid
Amyloids are aggregates of proteins characterised by a fibrillar morphology of 7–13 nm in diameter, a β-sheet secondary structure (known as cross-β) and ability to be stained by particular dyes, such as Congo red.[1] In the human body, amyloids have been linked to the development of various diseases.[2] Pathogenic amyloids form when previously healthy proteins lose their normal structure and physiological functions (misfolding) and form fibrous deposits in plaques around cells which can disrupt the healthy function of tissues and organs.
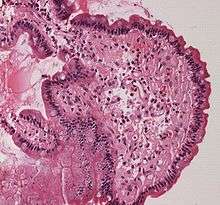
Such amyloids have been associated with (but not necessarily as the cause of) more than 50[2][3] human diseases, known as amyloidosis, and may play a role in some neurodegenerative disorders.[2][4] Some of these diseases are mainly sporadic and only a few cases are familial. Others are only familial. Some are iatrogenic as they result from medical treatment. One amyloid protein is infectious and is called prion in which the infectious form can act as a template to convert other non-infectious proteins into infectious form.[5] Amyloids may also have normal biological functions; for example, in the formation of fimbriae in some genera of bacteria, transmission of epigenetic traits in fungi, as well as pigment deposition and hormone release in humans.[6]
Amyloids have been known to arise from many different proteins.[2][7] These polypeptide chains generally form β-sheet structures that aggregate into long fibers; however, identical polypeptides can fold into multiple distinct amyloid conformations.[8] The diversity of the conformations may have led to different forms of the prion diseases.[6]
Definition
The name amyloid comes from the early mistaken identification by Rudolf Virchow of the substance as starch (amylum in Latin, from Greek ἄμυλον amylon), based on crude iodine-staining techniques. For a period, the scientific community debated whether or not amyloid deposits are fatty deposits or carbohydrate deposits until it was finally found (in 1859) that they are, in fact, deposits of albumoid proteinaceous material.[9]
- The classical, histopathological definition of amyloid is an extracellular, proteinaceous fibrillar deposit exhibiting β-sheet secondary structure and identified by apple-green birefringence when stained with congo red under polarized light. These deposits often recruit various sugars and other components such as Serum Amyloid P component, resulting in complex, and sometimes inhomogeneous structures.[10] Recently this definition has come into question as some classic, amyloid species have been observed in distinctly intracellular locations.[11]
- A more recent, biophysical definition is broader, including any polypeptide that polymerizes to form a cross-β structure, in vivo or in vitro, inside or outside cells. Microbiologists, biochemists, biophysicists, chemists and physicists have largely adopted this definition,[12][13] leading to some conflict in the biological community over an issue of language.
Proteins forming amyloids in diseases
To date, 37 human proteins have been found to form amyloid in pathology and be associated with well-defined diseases[2]. The International Society of Amyloidosis classifies amyloid fibrils and their associated diseases based upon associated proteins (for example ATTR is the group of diseases and associated fibrils formed by TTR).[3] A table is included below.
| Protein | Diseases | Official abbreviation |
|---|---|---|
| β amyloid peptide (Aβ) from Amyloid precursor protein[14][15][16][17] | Alzheimer's disease, Hereditary cerebral haemorrhage with amyloidosis | Aβ |
| α-synuclein[15] | Parkinson's disease, Parkinson's disease dementia, Dementia with Lewy bodies, Multiple system atrophy | AαSyn |
| PrPSc[18] | Transmissible spongiform encephalopathy (e.g. Fatal familial insomnia, Gerstmann-Sträussler-Scheinker disease, Creutzfeldt-Jacob disease, New variant Creutzfeldt-Jacob disease) | APrP |
| Microtubule-associated protein tau | Various forms of tauopathies (e.g. Pick’s disease, Progressive supranuclear palsy, Corticobasal degeneration, Frontotemporal dementia with parkinsonism linked to chromosome 17, Argyrophilic grain disease) | ATau |
| Huntingtin exon 1[19][20] | Huntington's disease | none |
| ABri peptide | Familial British dementia | ABri |
| ADan peptide | Familial Danish dementia | ADan |
| Fragments of immunoglobulin light chains[21] | Light chain amyloidosis | AL |
| Fragments of immunoglobulin heavy chains[21] | Heavy chain amyloidosis | AH |
| full length of N-terminal fragments of Serum amyloid A protein | AA amyloidosis | AA |
| Transthyretin | Senile systemic amyloidosis, Familial amyloid polyneuropathy, Familial amyloid cardiomyopathy, Leptomeningeal amyloidosis | ATTR |
| Beta-2 microglobulin | Dialysis related amyloidosis, Hereditary visceral amyloidosis (familial) | Aβ2M |
| N-terminal fragments of Apolipoprotein AI | ApoAI amyloidosis | AApoAI |
| C-terminally extended Apolipoprotein AII | ApoAII amyloidosis | AApoAII |
| N-terminal fragments of Apolipoprotein AIV | ApoAIV amyloidosis | AApoAIV |
| Apolipoprotein C-II | ApoCII amyloidosis | AApoCII |
| Apolipoprotein C-III | ApoCIII amyloidosis | AApoCIII |
| fragments of Gelsolin | familial amyloidosis, Finnish type | AGel |
| Lysozyme | Hereditary non-neuropathic systemic amyloidosis | ALys |
| fragments of Fibrinogen alpha chain | Fibrinogen amyloidosis | AFib |
| N-terminally truncated Cystatin C | Hereditary cerebral hemorrhage with amyloidosis, Icelandic type | ACys |
| IAPP (Amylin)[22][23] | Diabetes mellitus type 2, Insulinoma | AIAPP |
| Calcitonin[21] | Medullary carcinoma of the thyroid | ACal |
| Atrial natriuretic factor | Cardiac arrhythmias, Isolated atrial amyloidosis | AANF |
| Prolactin | Pituitary Prolactinoma | APro |
| Insulin | Injection-localized amyloidosis | AIns |
| Lactadherin / Medin | Aortic medial amyloidosis | AMed |
| Lactotransferrin / Lactoferrin | Gelatinous drop-like corneal dystrophy | ALac |
| Odontogenic ameloblast-associated protein | Calcifying epithelial odontogenic tumors | AOAAP |
| Pulmonary surfactant-associated protein C (SP-C) | Pulmonary alveolar proteinosis | ASPC |
| Leukocyte cell-derived chemotaxin-2 (LECT-2) | Renal LECT2 amyloidosis | ALECT2 |
| Galectin-7 | Lichen amyloidosis, Macular amyloidosis | AGal7 |
| Corneodesmosin | Hypotrichosis simplex of the scalp | ACor |
| C-terminal fragments of TGFBI/Keratoepithelin | Lattice corneal dystrophy type I, Lattice corneal dystrophy type 3A, Lattice corneal dystrophy Avellino type | AKer |
| Semenogelin-1 (SGI) | Seminal vesicle amyloidosis | ASem1 |
| Proteins S100A8/A9 | Prostate cancer | none |
| Enfuvirtide | Injection-localized amyloidosis | AEnf |
Non-disease and functional amyloids
Many examples of non-pathological amyloid with a well-defined physiological role have been identified in various organisms, including human. These may be termed as functional or physiological or native amyloid.[24][25][2]
- Functional amyloid in Homo sapiens:
- Intralumenal domain of melanocyte protein PMEL[26]
- Peptide/protein hormones stored as amyloids within endocrine secretory granules[27]
- Receptor-interacting serine/threonine-protein kinase 1/3 (RIP1/RIP3)[28]
- Fragments of prostatic acid phosphatase and semenogelins[29]
- Functional amyloid in other organisms:
- Curli fibrils produced by E. coli, Salmonella, and a few other members of the Enterobacteriales (Csg). The genetic elements (operons) encoding the curli system are phylogenetic widespread and can be found in at least four bacterial phyla.[30] This suggest that many more bacteria may express curli fibrils.
- GvpA, forming the walls of particular Gas vesicles, i.e. the buoyancy organelles of aquatic archaea and eubacteria[31]
- Fap fibrils in various species of Pseudomonas[32][33]
- Chaplins from Streptomyces coelicolor[34]
- Spidroin from Trichonephila edulis (spider) (Spider silk)[35]
- Hydrophobins from Neurospora crassa and other fungi[36]
- Fungal cell adhesion proteins forming cell surface amyloid regions with greatly increased binding strength [37][38]
- Environmental biofilms according to staining with amyloid specific dyes and antibodies.[39] For example, Bacillus subtilis biofilms involve the TasA protein, which forms functional amyloids maintaining biofilm structure.[40]
- Tubular sheaths encasing Methanosaeta thermophila filaments[41]
- Functional amyloid acting as prions
- Several yeast prions are based on an infectious amyloid, e.g. [PSI+] (Sup35p); [URE3] (Ure2p); [PIN+] or [RNQ+] (Rnq1p); [SWI1+] (Swi1p) and [OCT8+] (Cyc8p)
- Prion HET-s from Podospora anserina[42]
- Neuron-specific isoform of CPEB from Aplysia Californica (marine snail)[43]
Structure
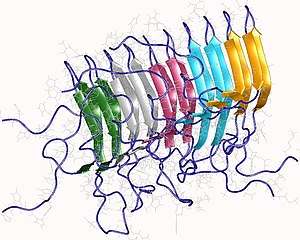
Amyloids are formed of long unbranched fibers that are characterized by an extended beta-sheet secondary structure in which individual β-strands (coloured arrows in the figure on the right) are arranged in an orientation perpendicular to the axis of the fiber. Such a structure is known as cross-β structure. Each individual fiber may be 5–15 nanometres in width and a few micrometres in length.[6][2] The main hallmarks recognised by different disciplines to classify protein aggregates as amyloid is the presence of a fibrillar morphology with the expected diameter, detected using transmission electron microscopy (TEM) or atomic force microscopy (AFM), the presence of a cross-β secondary structure, determined with circular dichroism, FTIR, solid-state nuclear magnetic resonance (ssNMR), X-ray crystallography, or X-ray fiber diffraction (often considered the "gold-standard" test to see whether a structure contains cross-β fibres), and an ability to stain with specific dyes, such as Congo red, thioflavin T or thioflavin S.[2]
The term "cross-β" was based on the observation of two sets of diffraction lines, one longitudinal and one transverse, that form a characteristic "cross" pattern.[44] There are two characteristic scattering diffraction signals produced at 4.7 and 10 Ångstroms (0.47 nm and 1.0 nm), corresponding to the interstrand and stacking distances in beta sheets.[1] The "stacks" of beta sheet are short and traverse the breadth of the amyloid fibril; the length of the amyloid fibril is built by aligned β-strands. The cross-β pattern is considered a diagnostic hallmark of amyloid structure.[6]
Amyloid fibrils are generally composed of 2–8 protofilaments (four are shown in the figure), each 2–7 nm in diameter, that interact laterally as flat ribbons that maintain the height of 2–7 nm (that of a single protofilament) and are 30 nm wide; more often protofilaments twist around each other to form the typically 5–15 nm wide fibrils.[2] Each protofilament possesses the typical cross-β structure and may be formed by 1–4 β-sheets (two are shown in the figure) stacked on each other. Each individual protein molecule can contribute one to several β-strands in each protofilament and the strands can be arranged in antiparallel β-sheets, but more often in parallel β-sheets. Only a fraction of the polypeptide chain is in a β-strand conformation in the fibrils, the remainder forms structured or unstructured loops or tails.
For a long time our knowledge of the atomic-level structure of amyloid fibrils was limited by the fact that they are unsuitable for the most traditional methods for studying protein structures. Recent years have seen progress in experimental methods, including solid-state NMR spectroscopy and Cryo-Electron Microscopy. Combined, these methods have provided 3D atomic structures of amyloid fibrils formed by amyloid β peptides, α-synuclein, tau, and the FUS protein, associated with various neurodegenerative diseases.[45][46]
X-ray diffraction studies of microcrystals revealed atomistic details of core region of amyloid, although only for simplified peptides having a length remarkably shorter than that of peptides or proteins involved in disease.[47][48] The crystallographic structures show that short stretches from amyloid-prone regions of amyloidogenic proteins run perpendicular to the filament axis, consistent with the "cross-β" feature of amyloid structure. They also reveal a number of characteristics of amyloid structures – neighboring β-sheets are tightly packed together via an interface devoid of water (therefore referred to as dry interface), with the opposing β-strands slightly offset from each other such that their side-chains interdigitate. This compact dehydrated interface created was termed a steric-zipper interface.[6] There are eight theoretical classes of steric-zipper interfaces, dictated by the directionality of the β-sheets (parallel and anti-parallel) and symmetry between adjacent β-sheets. A limitation of X-ray crystallography for solving amyloid structure is represented by the need to form microcrystals, which can be achieved only with peptides shorter than those associated with disease.
Although bona fide amyloid structures always are based on intermolecular β-sheets, different types of "higher order" tertiary folds have been observed or proposed. The β-sheets may form a β-sandwich, or a β-solenoid which may be either β-helix or β-roll. Native-like amyloid fibrils in which native β-sheet containing proteins maintain their native-like structure in the fibrils have also been proposed.[49]
One complicating factor in studies of amyloidogenic polypeptides is that identical polypeptides can fold into multiple distinct amyloid conformations.[6] This phenomenon is typically described as amyloid polymorphism.[8][50] [51] It has notable biological consequences given that it is thought to explain the prion strain phenomenon.
Formation
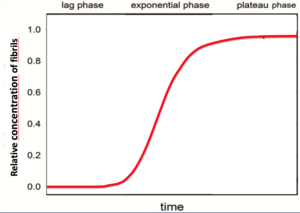
Amyloid is formed through the polymerization of hundreds to thousands of monomeric peptides or proteins into long fibers. Amyloid formation involves a lag phase (also called nucleation phase), an exponential phase (also called growth phase) and a plateau phase (also called saturation phase), as shown in the figure.[52][53][54][55] Indeed, when the quantity of fibrils is plotted versus time, a sigmoidal time course is observed reflecting the three distinct phases.
In the simplest model of ‘nucleated polymerization’ (marked by red arrows in the figure below), individual unfolded or partially unfolded polypeptide chains (monomers) convert into a nucleus (monomer or oligomer) via a thermodynamically unfavourable process that occurs early in the lag phase.[54] Fibrils grow subsequently from these nuclei through the addition of monomers in the exponential phase.[54]
A different model, called ‘nucleated conformational conversion’ and marked by blue arrows in the figure below, was introduced later on to fit some experimental observations: monomers have often been found to convert rapidly into misfolded and highly disorganized oligomers distinct from nuclei.[56] Only later on, will these aggregates reorganise structurally into nuclei, on which other disorganised oligomers will add and reorganise through a templating or induced-fit mechanism (this ‘nucleated conformational conversion’ model), eventually forming fibrils.[56]
Normally folded proteins have to unfold partially before aggregation can take place through one of these mechanisms.[57] In some cases, however, folded proteins can aggregate without crossing the major energy barrier for unfolding, by populating native-like conformations as a consequence of thermal fluctuations, ligand release or local unfolding occurring in particular circumstances.[57] In these native-like conformations, segments that are normally buried or structured in the fully folded and possessing a high propensity to aggregate become exposed to the solvent or flexible, allowing the formation of native-like aggregates, which convert subsequently into nuclei and fibrils. This process is called ‘native-like aggregation’ (green arrows in the figure) and is similar to the ‘nucleated conformational conversion’ model.
A more recent, modern and thorough model of amyloid fibril formation involves the intervention of secondary events, such as ‘fragmentation’, in which a fibril breaks into two or more shorter fibrils, and ‘secondary nucleation’, in which fibril surfaces (not fibril ends) catalyze the formation of new nuclei.[55] Both secondary events increase the number of fibril ends able to recruit new monomers or oligomers, therefore accelerating fibril formation. These events add to the well recognised steps of primary nucleation (formation of the nucleus from the mnonomers through one of models described above), fibril elongation (addition of monomers or oligomers to growing fibril ends) and dissociation (opposite process).
Such a new model is described in the figure on the right and involves the utilization of a ‘master equation’ that includes all steps of amyloid fibril formation, i.e. primary nucleation, fibril elongation, secondary nucleation and fibril fragmentation.[55] The rate constants of the various steps can be determined from a global fit of a number of time courses of aggregation (for example ThT fluorescence emission versus time) recorded at different protein concentrations.[55]
Following this analytical approach, it has become apparent that the lag phase does not correspond necessarily to only nucleus formation, but rather results from a combination of various steps. Similarly, the exponential phase is not only fibril elongation, but results from a combination of various steps, involving primary nucleation, fibril elongation, but also secondary events. A significant quantity of fibrils resulting from primary nucleation and fibril elongation may be formed during the lag phase and secondary steps, rather than only fibril elongation, can be the dominant processes contributing to fibril growth during the exponential phase. With this new model, any perturbing agents of amyloid fibril formation, such as putative drugs, metabolites, mutations, chaperones, etc., can be assigned to a specific step of fibril formation.
Amino acid sequence and amyloid formation
In general, amyloid polymerization (aggregation or non-covalent polymerization) is sequence-sensitive, that is mutations in the sequence can induce or prevent self-assembly.[58][59] For example, humans produce amylin, an amyloidogenic peptide associated with type II diabetes, but in rats and mice prolines are substituted in critical locations and amyloidogenesis does not occur. Studies comparing synthetic to recombinant β amyloid peptide in assays measuring rate of fibrillation, fibril homogeneity, and cellular toxicity showed that recombinant β amyloid peptide has a faster fibrillation rate and greater toxicity than synthetic β amyloid peptide.[60]
There are multiple classes of amyloid-forming polypeptide sequences.[8][50][51] Glutamine-rich polypeptides are important in the amyloidogenesis of Yeast and mammalian prions, as well as Trinucleotide repeat disorders including Huntington's disease. When glutamine-rich polypeptides are in a β-sheet conformation, glutamines can brace the structure by forming inter-strand hydrogen bonding between its amide carbonyls and nitrogens of both the backbone and side chains. The onset age for Huntington's disease shows an inverse correlation with the length of the polyglutamine sequence, with analogous findings in a C. elegans model system with engineered polyglutamine peptides.[61]
Other polypeptides and proteins such as amylin and the β amyloid peptide do not have a simple consensus sequence and are thought to aggregate through the sequence segments enriched with hydrophobic residues, or residues with high propensity to form β-sheet structure.[58] Among the hydrophobic residues, aromatic amino-acids are found to have the highest amyloidogenic propensity.[62][63]
Cross-polymerization (fibrils of one polypeptide sequence causing other fibrils of another sequence to form) is observed in vitro and possibly in vivo. This phenomenon is important, since it would explain interspecies prion propagation and differential rates of prion propagation, as well as a statistical link between Alzheimer's and type 2 diabetes.[64] In general, the more similar the peptide sequence the more efficient cross-polymerization is, though entirely dissimilar sequences can cross-polymerize and highly similar sequences can even be "blockers" that prevent polymerization.
Amyloid toxicity
The reasons why amyloid cause diseases are unclear. In some cases, the deposits physically disrupt tissue architecture, suggesting disruption of function by some bulk process. An emerging consensus implicates prefibrillar intermediates, rather than mature amyloid fibers, in causing cell death, particularly in neurodegenerative diseases.[16][65] The fibrils are, however, far from innocuous, as they keep the protein homeostasis network engaged, release oligomers, cause the formation of toxic oligomers via secondary nucleation, grow indefinitely spreading from district to district[2] and, in some cases, may be toxic themselves.[66]
Calcium dysregulation has been observed to occur early in cells exposed to protein oligomers. These small aggregates can form ion channels through lipid bilayer membranes and activate NMDA and AMPA receptors. Channel formation has been hypothesized to account for calcium dysregulation and mitochondrial dysfunction by allowing indiscriminate leakage of ions across cell membranes.[67] Studies have shown that amyloid deposition is associated with mitochondrial dysfunction and a resulting generation of reactive oxygen species (ROS), which can initiate a signalling pathway leading to apoptosis.[68] There are reports that indicate amyloid polymers (such as those of huntingtin, associated with Huntington's disease) can induce the polymerization of essential amyloidogenic proteins, which should be deleterious to cells. Also, interaction partners of these essential proteins can also be sequestered.[69]
All these mechanisms of toxicity are likely to play a role. In fact, the aggregation of a protein generates a variety of aggregates, all of which are likely to be toxic to some degree. A wide variety of biochemical, physiological and cytological perturbations has been identified following the exposure of cells and animals to such species, independently of their identity. The oligomers have also been reported to interact with a variety of molecular targets. Hence, it is unlikely that there is a unique mechanism of toxicity or a unique cascade of cellular events. The misfolded nature of protein aggregates causes a multitude of aberrant interactions with a multitude of cellular components, including membranes, protein receptors, soluble proteins, RNAs, small metabolites, etc.
Histological staining
In the clinical setting, amyloid diseases are typically identified by a change in the spectroscopic properties of planar aromatic dyes such as thioflavin T, congo red or NIAD-4.[70] In general, this is attributed to the environmental change, as these dyes intercalate between beta-strands to confine their structure.[71]
Congo Red positivity remains the gold standard for diagnosis of amyloidosis. In general, binding of Congo Red to amyloid plaques produces a typical apple-green birefringence when viewed under cross-polarized light. Recently, significant enhancement of fluorescence quantum yield of NIAD-4 was exploited to super-resolution fluorescence imaging of amyloid fibrils[72] and oligomers.[73] To avoid nonspecific staining, other histology stains, such as the hematoxylin and eosin stain, are used to quench the dyes' activity in other places such as the nucleus, where the dye might bind. Modern antibody technology and immunohistochemistry has made specific staining easier, but often this can cause trouble because epitopes can be concealed in the amyloid fold; in general, an amyloid protein structure is a different conformation from the one that the antibody recognizes.
See also
References
- Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC (October 1997). "Common core structure of amyloid fibrils by synchrotron X-ray diffraction". Journal of Molecular Biology. 273 (3): 729–39. doi:10.1006/jmbi.1997.1348. PMID 9356260. S2CID 19394482.
- Chiti F, Dobson CM (June 2017). "Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade". Annual Review of Biochemistry. 86: 27–68. doi:10.1146/annurev-biochem-061516-045115. PMID 28498720.
- Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJ, Sekijima Y, et al. (December 2018). "Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee". Amyloid. 25 (4): 215–219. doi:10.1080/13506129.2018.1549825. PMID 30614283.
- Pulawski W, Ghoshdastider U, Andrisano V, Filipek S (April 2012). "Ubiquitous amyloids". Applied Biochemistry and Biotechnology. 166 (7): 1626–43. doi:10.1007/s12010-012-9549-3. PMC 3324686. PMID 22350870.
- Soto C, Estrada L, Castilla J (March 2006). "Amyloids, prions and the inherent infectious nature of misfolded protein aggregates". Trends in Biochemical Sciences. 31 (3): 150–5. doi:10.1016/j.tibs.2006.01.002. PMID 16473510.
- Toyama BH, Weissman JS (2011). "Amyloid structure: conformational diversity and consequences". Annual Review of Biochemistry. 80: 557–85. doi:10.1146/annurev-biochem-090908-120656. PMC 3817101. PMID 21456964.
- Ramirez-Alvarado M, Merkel JS, Regan L (August 2000). "A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro". Proceedings of the National Academy of Sciences of the United States of America. 97 (16): 8979–84. Bibcode:2000PNAS...97.8979R. doi:10.1073/pnas.150091797. PMC 16807. PMID 10908649.
- Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, et al. (November 2000). "Amyloid fibril formation by A beta 16-22, a seven-residue fragment of the Alzheimer's beta-amyloid peptide, and structural characterization by solid state NMR". Biochemistry. 39 (45): 13748–59. doi:10.1021/bi0011330. PMID 11076514. S2CID 17232045.
- Kyle RA (September 2001). "Amyloidosis: a convoluted story". British Journal of Haematology. 114 (3): 529–38. doi:10.1046/j.1365-2141.2001.02999.x. PMID 11552976.
- Sipe JD, Cohen AS (June 2000). "Review: history of the amyloid fibril". Journal of Structural Biology. 130 (2–3): 88–98. doi:10.1006/jsbi.2000.4221. PMID 10940217. S2CID 16442783.
- Lin CY, Gurlo T, Kayed R, Butler AE, Haataja L, Glabe CG, Butler PC (May 2007). "Toxic human islet amyloid polypeptide (h-IAPP) oligomers are intracellular, and vaccination to induce anti-toxic oligomer antibodies does not prevent h-IAPP-induced beta-cell apoptosis in h-IAPP transgenic mice". Diabetes. 56 (5): 1324–32. doi:10.2337/db06-1579. PMID 17353506.
- Nilsson MR (September 2004). "Techniques to study amyloid fibril formation in vitro". Methods. 34 (1): 151–60. doi:10.1016/j.ymeth.2004.03.012. PMID 15283924.
- Fändrich M (August 2007). "On the structural definition of amyloid fibrils and other polypeptide aggregates". Cellular and Molecular Life Sciences. 64 (16): 2066–78. doi:10.1007/s00018-007-7110-2. PMID 17530168.
- Chiang PK, Lam MA, Luo Y (September 2008). "The many faces of amyloid beta in Alzheimer's disease". Current Molecular Medicine. 8 (6): 580–4. doi:10.2174/156652408785747951. PMID 18781964.
- Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM (2008). "Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases". Molecular Medicine. 14 (7–8): 451–64. doi:10.2119/2007-00100.Irvine. PMC 2274891. PMID 18368143.
- Ferreira ST, Vieira MN, De Felice FG (2007). "Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases". IUBMB Life. 59 (4–5): 332–45. doi:10.1080/15216540701283882. PMID 17505973. S2CID 7489461.
- Hamley IW (October 2012). "The amyloid beta peptide: a chemist's perspective. Role in Alzheimer's and fibrillization" (PDF). Chemical Reviews. 112 (10): 5147–92. doi:10.1021/cr3000994. PMID 22813427.
- "More than just mad cow disease". Nature Structural Biology. 8 (4): 281. April 2001. doi:10.1038/86132. PMID 11276238.
- Truant R, Atwal RS, Desmond C, Munsie L, Tran T (September 2008). "Huntington's disease: revisiting the aggregation hypothesis in polyglutamine neurodegenerative diseases". The FEBS Journal. 275 (17): 4252–62. doi:10.1111/j.1742-4658.2008.06561.x. PMID 18637947.
- Weydt P, La Spada AR (August 2006). "Targeting protein aggregation in neurodegeneration--lessons from polyglutamine disorders". Expert Opinion on Therapeutic Targets. 10 (4): 505–13. doi:10.1517/14728222.10.4.505. PMID 16848688.
- Holmes RO, Edison J, Baethge BA, Jacobson DR (10 October 2018). "Amyloidosis: Definition of Amyloid and Amyloidosis, Classification Systems, Systemic Amyloidoses". Medscape.
- Haataja L, Gurlo T, Huang CJ, Butler PC (May 2008). "Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis". Endocrine Reviews. 29 (3): 303–16. doi:10.1210/er.2007-0037. PMC 2528855. PMID 18314421.
- Höppener JW, Ahrén B, Lips CJ (August 2000). "Islet amyloid and type 2 diabetes mellitus". The New England Journal of Medicine. 343 (6): 411–9. doi:10.1056/NEJM200008103430607. PMID 10933741.
- Hammer ND, Wang X, McGuffie BA, Chapman MR (May 2008). "Amyloids: friend or foe?". Journal of Alzheimer's Disease. 13 (4): 407–19. doi:10.3233/JAD-2008-13406. PMC 2674399. PMID 18487849. Archived from the original on 2013-01-03.
- Fowler DM, Koulov AV, Balch WE, Kelly JW (May 2007). "Functional amyloid--from bacteria to humans". Trends in Biochemical Sciences. 32 (5): 217–24. doi:10.1016/j.tibs.2007.03.003. PMID 17412596.
- Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW (January 2006). "Functional amyloid formation within mammalian tissue". PLOS Biology. 4 (1): e6. doi:10.1371/journal.pbio.0040006. PMC 1288039. PMID 16300414.
- Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, et al. (July 2009). "Functional amyloids as natural storage of peptide hormones in pituitary secretory granules". Science. 325 (5938): 328–32. Bibcode:2009Sci...325..328M. doi:10.1126/science.1173155. PMC 2865899. PMID 19541956.
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. (July 2012). "The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis". Cell. 150 (2): 339–50. doi:10.1016/j.cell.2012.06.019. PMC 3664196. PMID 22817896.
- Usmani SM, Zirafi O, Müller JA, Sandi-Monroy NL, Yadav JK, Meier C, et al. (April 2014). "Direct visualization of HIV-enhancing endogenous amyloid fibrils in human semen". Nature Communications. 5: 3508. Bibcode:2014NatCo...5.3508U. doi:10.1038/ncomms4508. PMC 4129123. PMID 24691351.
- Dueholm MS, Albertsen M, Otzen D, Nielsen PH (2012). Webber MA (ed.). "Curli functional amyloid systems are phylogenetically widespread and display large diversity in operon and protein structure". PLOS ONE. 7 (12): e51274. Bibcode:2012PLoSO...751274D. doi:10.1371/journal.pone.0051274. PMC 3521004. PMID 23251478.
- Bayro MJ, Daviso E, Belenky M, Griffin RG, Herzfeld J (January 2012). "An amyloid organelle, solid-state NMR evidence for cross-β assembly of gas vesicles". The Journal of Biological Chemistry. 287 (5): 3479–84. doi:10.1074/jbc.M111.313049. PMC 3271001. PMID 22147705.
- Dueholm MS, Petersen SV, Sønderkær M, Larsen P, Christiansen G, Hein KL, et al. (August 2010). "Functional amyloid in Pseudomonas". Molecular Microbiology. 77 (4): 1009–20. doi:10.1111/j.1365-2958.2010.07269.x. PMID 20572935.
- Dueholm MS, Søndergaard MT, Nilsson M, Christiansen G, Stensballe A, Overgaard MT, et al. (June 2013). "Expression of Fap amyloids in Pseudomonas aeruginosa, P. fluorescens, and P. putida results in aggregation and increased biofilm formation". MicrobiologyOpen. 2 (3): 365–82. doi:10.1002/mbo3.81. PMC 3684753. PMID 23504942.
- Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, Boersma FG, et al. (July 2003). "A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils". Genes & Development. 17 (14): 1714–26. doi:10.1101/gad.264303. PMC 196180. PMID 12832396.
- Kenney JM, Knight D, Wise MJ, Vollrath F (August 2002). "Amyloidogenic nature of spider silk". European Journal of Biochemistry. 269 (16): 4159–63. doi:10.1046/j.1432-1033.2002.03112.x. PMID 12180993.
- Mackay JP, Matthews JM, Winefield RD, Mackay LG, Haverkamp RG, Templeton MD (February 2001). "The hydrophobin EAS is largely unstructured in solution and functions by forming amyloid-like structures". Structure. 9 (2): 83–91. doi:10.1016/s0969-2126(00)00559-1. PMID 11250193.
- Garcia MC, Lee JT, Ramsook CB, Alsteens D, Dufrêne YF, Lipke PN (March 2011). "A role for amyloid in cell aggregation and biofilm formation". PLOS ONE. 6 (3): e17632. Bibcode:2011PLoSO...617632G. doi:10.1371/journal.pone.0017632. PMC 3050909. PMID 21408122.
- Lipke PN, Garcia MC, Alsteens D, Ramsook CB, Klotz SA, Dufrêne YF (February 2012). "Strengthening relationships: amyloids create adhesion nanodomains in yeasts". Trends in Microbiology. 20 (2): 59–65. doi:10.1016/j.tim.2011.10.002. PMC 3278544. PMID 22099004.
- Larsen P, Nielsen JL, Dueholm MS, Wetzel R, Otzen D, Nielsen PH (December 2007). "Amyloid adhesins are abundant in natural biofilms". Environmental Microbiology. 9 (12): 3077–90. doi:10.1111/j.1462-2920.2007.01418.x. PMID 17991035.
- Kovács AT, van Gestel J, Kuipers OP (July 2012). "The protective layer of biofilm: a repellent function for a new class of amphiphilic proteins" (PDF). Molecular Microbiology. 85 (1): 8–11. doi:10.1111/j.1365-2958.2012.08101.x. PMID 22607588.
- Dueholm MS, Larsen P, Finster K, Stenvang MR, Christiansen G, Vad BS, et al. (August 2015). "The Tubular Sheaths Encasing Methanosaeta thermophila Filaments Are Functional Amyloids". The Journal of Biological Chemistry. 290 (33): 20590–600. doi:10.1074/jbc.M115.654780. PMC 4536462. PMID 26109065.
- Coustou V, Deleu C, Saupe S, Begueret J (September 1997). "The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog". Proceedings of the National Academy of Sciences of the United States of America. 94 (18): 9773–8. Bibcode:1997PNAS...94.9773C. doi:10.1073/pnas.94.18.9773. PMC 23266. PMID 9275200.
- Si K, Lindquist S, Kandel ER (December 2003). "A neuronal isoform of the aplysia CPEB has prion-like properties". Cell. 115 (7): 879–91. doi:10.1016/s0092-8674(03)01020-1. PMID 14697205.
- Wormell RL. New fibres from proteins. Academic Press, 1954, p. 106.
- Meier BH, Riek R, Böckmann A (October 2017). "Emerging Structural Understanding of Amyloid Fibrils by Solid-State NMR". Trends in Biochemical Sciences. 42 (10): 777–787. doi:10.1016/j.tibs.2017.08.001. hdl:20.500.11850/193533. PMID 28916413.
- Fitzpatrick AW, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. (July 2017). "Cryo-EM structures of tau filaments from Alzheimer's disease". Nature. 547 (7662): 185–190. Bibcode:2017Natur.547..185F. doi:10.1038/nature23002. PMC 5552202. PMID 28678775.
- Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, Eisenberg D (June 2005). "Structure of the cross-beta spine of amyloid-like fibrils". Nature. 435 (7043): 773–8. Bibcode:2005Natur.435..773N. doi:10.1038/nature03680. PMC 1479801. PMID 15944695.
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, et al. (May 2007). "Atomic structures of amyloid cross-beta spines reveal varied steric zippers". Nature. 447 (7143): 453–7. Bibcode:2007Natur.447..453S. doi:10.1038/nature05695. PMID 17468747.
- Serag AA, Altenbach C, Gingery M, Hubbell WL, Yeates TO (October 2002). "Arrangement of subunits and ordering of beta-strands in an amyloid sheet". Nature Structural Biology. 9 (10): 734–9. doi:10.1038/nsb838. PMID 12219081.
- Bu Z, Shi Y, Callaway DJ, Tycko R (January 2007). "Molecular alignment within beta-sheets in Abeta(14-23) fibrils: solid-state NMR experiments and theoretical predictions". Biophysical Journal. 92 (2): 594–602. doi:10.1529/biophysj.106.091017. PMC 1751388. PMID 17056725.
- Tjernberg LO, Tjernberg A, Bark N, Shi Y, Ruzsicska BP, Bu Z, et al. (August 2002). "Assembling amyloid fibrils from designed structures containing a significant amyloid beta-peptide fragment". The Biochemical Journal. 366 (Pt 1): 343–51. doi:10.1042/BJ20020229. PMC 1222771. PMID 12023906.
- Jarrett JT, Berger EP, Lansbury PT (May 1993). "The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease". Biochemistry. 32 (18): 4693–7. doi:10.1021/bi00069a001. PMID 8490014.
- Ferrone F (1999). "Analysis of protein aggregation kinetics". Methods in Enzymology. 309: 256–74. doi:10.1016/s0076-6879(99)09019-9. PMID 10507029.
- Morris AM, Watzky MA, Finke RG (March 2009). "Protein aggregation kinetics, mechanism, and curve-fitting: a review of the literature". Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 1794 (3): 375–97. doi:10.1016/j.bbapap.2008.10.016. PMID 19071235.
- Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, et al. (December 2009). "An analytical solution to the kinetics of breakable filament assembly". Science. 326 (5959): 1533–7. Bibcode:2009Sci...326.1533K. doi:10.1126/science.1178250. PMID 20007899. S2CID 6267152.
- Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, et al. (August 2000). "Nucleated conformational conversion and the replication of conformational information by a prion determinant". Science. 289 (5483): 1317–21. Bibcode:2000Sci...289.1317S. doi:10.1126/science.289.5483.1317. PMID 10958771.
- Chiti F, Dobson CM (January 2009). "Amyloid formation by globular proteins under native conditions". Nature Chemical Biology. 5 (1): 15–22. doi:10.1038/nchembio.131. PMID 19088715.
- Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM (August 2003). "Rationalization of the effects of mutations on peptide and protein aggregation rates". Nature. 424 (6950): 805–8. Bibcode:2003Natur.424..805C. doi:10.1038/nature01891. PMID 12917692.
- Gilead S, Gazit E (August 2004). "Inhibition of amyloid fibril formation by peptide analogues modified with alpha-aminoisobutyric acid". Angewandte Chemie. 43 (31): 4041–4. doi:10.1002/anie.200353565. PMID 15300690.
- Finder VH, Vodopivec I, Nitsch RM, Glockshuber R (February 2010). "The recombinant amyloid-beta peptide Abeta1-42 aggregates faster and is more neurotoxic than synthetic Abeta1-42". Journal of Molecular Biology. 396 (1): 9–18. doi:10.1016/j.jmb.2009.12.016. PMID 20026079.
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI (August 2002). "The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans". Proceedings of the National Academy of Sciences of the United States of America. 99 (16): 10417–22. Bibcode:2002PNAS...9910417M. doi:10.1073/pnas.152161099. PMC 124929. PMID 12122205.
- Gazit E (January 2002). "A possible role for pi-stacking in the self-assembly of amyloid fibrils". FASEB Journal. 16 (1): 77–83. doi:10.1096/fj.01-0442hyp. PMID 11772939.
- Pawar AP, Dubay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM (July 2005). "Prediction of "aggregation-prone" and "aggregation-susceptible" regions in proteins associated with neurodegenerative diseases". Journal of Molecular Biology. 350 (2): 379–92. doi:10.1016/j.jmb.2005.04.016. PMID 15925383.
- Jackson K, Barisone GA, Diaz E, Jin LW, DeCarli C, Despa F (October 2013). "Amylin deposition in the brain: A second amyloid in Alzheimer disease?". Annals of Neurology. 74 (4): 517–26. doi:10.1002/ana.23956. PMC 3818462. PMID 23794448.
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG (April 2005). "Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers". The Journal of Biological Chemistry. 280 (17): 17294–300. doi:10.1074/jbc.M500997200. PMID 15722360.
- Gath J, Bousset L, Habenstein B, Melki R, Böckmann A, Meier BH (March 5, 2014). "Unlike twins: an NMR comparison of two α-synuclein polymorphs featuring different toxicity". PLOS ONE. 9 (3): e90659. Bibcode:2014PLoSO...990659G. doi:10.1371/journal.pone.0090659. PMC 3944079. PMID 24599158.
- Kagan BL, Azimov R, Azimova R (November 2004). "Amyloid peptide channels". The Journal of Membrane Biology. 202 (1): 1–10. doi:10.1007/s00232-004-0709-4. PMID 15702375.
- Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K, et al. (January 2005). "Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation". Cell Death and Differentiation. 12 (1): 19–24. doi:10.1038/sj.cdd.4401528. PMID 15592360.
- Kochneva-Pervukhova NV, Alexandrov AI, Ter-Avanesyan MD (2012). Tuite MF (ed.). "Amyloid-mediated sequestration of essential proteins contributes to mutant huntingtin toxicity in yeast". PLOS ONE. 7 (1): e29832. Bibcode:2012PLoSO...729832K. doi:10.1371/journal.pone.0029832. PMC 3256205. PMID 22253794.
- Nesterov EE, Skoch J, Hyman BT, Klunk WE, Bacskai BJ, Swager TM (August 2005). "In vivo optical imaging of amyloid aggregates in brain: design of fluorescent markers". Angewandte Chemie. 44 (34): 5452–6. doi:10.1002/anie.200500845. PMID 16059955. S2CID 42217289.
- Bae S, Lim E, Hwang D, Huh H, Kim SK (2015). "Torsion-dependent fluorescence switching of amyloid-binding dye NIAD-4". Chemical Physics Letters. 633: 109–13. Bibcode:2015CPL...633..109B. doi:10.1016/j.cplett.2015.05.010.
- Ries J, Udayar V, Soragni A, Hornemann S, Nilsson KP, Riek R, et al. (July 2013). "Superresolution imaging of amyloid fibrils with binding-activated probes". ACS Chemical Neuroscience. 4 (7): 1057–61. doi:10.1021/cn400091m. PMC 3715833. PMID 23594172.
- Huh H, Lee J, Kim HJ, Hohng S, Kim SK (2017). "Morphological analysis of oligomeric vs. fibrillar forms of α-synuclein aggregates with super-resolution BALM imaging". Chemical Physics Letters. 690: 62–67. Bibcode:2017CPL...690...62H. doi:10.1016/j.cplett.2017.10.034.
External links
- Bacterial Inclusion Bodies Contain Amyloid-Like Structure at SciVee
- Amyloid Cascade Hypothesis
- Amyloid: Journal of Protein Folding Disorders web page at InformaWorld
- Information, support and advice to anyone with Amyloidosis, particularly in Australia
- Role of anesthetics in Alzheimer's disease: Molecular details revealed
| Wikimedia Commons has media related to Amyloid. |