Allylic strain
Allylic strain (also known as A1,3 strain, 1,3-allylic strain, or A-strain) in organic chemistry is a type of strain energy resulting from the interaction between a substituent on one end of an olefin with an allylic substituent on the other end.[1] If the substituents (R and R') are large enough in size, they can sterically interfere with each other such that one conformer is greatly favored over the other.[2] Allyic strain was first recognized in the literature in 1965 by Johnson and Malhotra. The authors were investigating cyclohexane conformations including endocyclic and exocylic double bonds when they noticed certain conformations were disfavored due to the geometry constraints caused by the double bond.[3] Organic chemists capitalize on the rigidity resulting from allylic strain for use in asymmetric reactions.[2]

Quantifying allylic strain energy
The "strain energy" of a molecule is a quantity that is difficult to precisely define, so the meaning of this term can easily vary depending on one's interpretation.[4] Instead, an objective way to view the allylic strain of a molecule is through its conformational equilibrium. Comparing the heats of formation of the involved conformers, an overall ΔHeq can be evaluated. This term gives information about the relative stabilities of the involved conformers and the effect allylic strain has on equilibrium. Heats of formation can be determined experimentally though calorimetric studies; however, calculated enthalpies are more commonly used due to the greater ease of acquisition.[4]
Different methods utizilized to estimate conformational equilibrium enthalpy include: the Westheimer method,[5] the homomorph method,[6] and more simply—using estimated enthalpies of nonbonded interactions within a molecule.[3] Because all of these methods are approximations, reported strain values for the same molecule can vary and should be used only to give a general idea of the strain energy.
Olefins
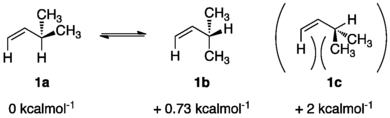
The simplest type of molecules which exhibit allylic strain are olefins. Depending on the substituents, olefins maintain varying degrees of allylic strain. In 3-methyl-1-butene, the interactions between the hydrogen and the two methyl groups in the allylic system cause a change in enthalpy equal to 2 kcal/mol.[7] As expected, with an increase in substituent size, the equilibrium enthalpies between rotamers also increases. For example, when examining 4-methyl-2-pentene which contains an additional allylic methyl group compared to 3-methyl-1-butene, the enthalpy of rotation for the highest energy conformer increases from 2 kcal/mol to 4 kcal/mol.[7]
Cyclic molecules
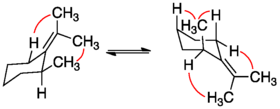
Nonbonded 1,3-diaxial interaction energies are commonly used to approximate strain energy in cyclic molecules, as values for these interactions are available. By taking the difference in nonbonded interactions for each conformer, the equilibrium enthalpy can be estimated. The strain energy for methylidenecyclohexane has been calculated to be 4.5 kcalmol−1 using estimations for 1,3-diaxial strain (0.9 kcalmol−1), methyl/hydrogen allylic strain (1.3kcalmol−1), and methyl/methyl allylic strain (7.6 kcalmol−1) values.[2]
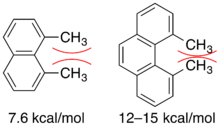
The strain energy in 1,8-dimethylnaphthalene was calculated to be 7.6 kcalmol−1 and around 12-15 kcalmol−1 for 4,5-dimethylphenanthrene.[2] Allylic strain tends to be greater for cyclic molecules compared to olefins as strain energy increases with increasing rigidity of the system. An in depth summary of allylic strain in six membered rings has been presented in a review by Johnson, F.[2]
Influencing factors
Several factors influence the energy penalty associated with the allylic strain. In order to relieve strain caused by interaction between the two methyl groups, the cyclohexanes will often exhibit a boat or twist-boat conformation. The boat conformation tends to be the major conformation to the strain.[2] The effect of allylic strain on cis alkenes creates a preference for more linear structures.[1]
Substituent size

The size of the substituents interacting at the 1 and 3 positions of an allylic group is often the largest factor contributing to the magnitude of the strain. As a rule, larger substituents will create a larger magnitude of strain. Proximity of bulky groups causes an increase in repulsive Van der Waals forces. This quickly increases the magnitude of the strain. The interactions between the hydrogen and methyl group in the allylic system cause a change in enthalpy equal to 3.6 kcal/mol.[7] The strain energy in this system was calculated to be 7.6 kcal/mol due to interactions between the two methyl groups.[2]
Substituent polarity
Polarity also has an effect on allylic strain. In terms of stereoselectivity, polar groups act like large, bulky groups. Even though two groups may have approximately the same A values the polar group will act as though it were much bulkier. This is due to the donor character of the polar group. Polar groups increase the HOMO energy of the σ-system in the transition state. This causes the transition state to be in a much more favorable position when the polar group is not interacting in a 1,3 allylic strain.[8]
Hydrogen bonding
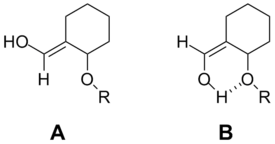
With certain polar substituents, hydrogen bonding can occur in the allylic system between the substituents. Rather than the strain that would normally occur in the close group proximity, the hydrogen bond stabilizes the conformation and makes it energetically much more favorable. This scenario occurs when the allylic substituent at the 1 position is a hydrogen bond donor (usually a hydroxyl) and the substituent at the 3 position is a hydrogen bond acceptor (usually an ether). Even in cases where the allylic system could conform to put a much smaller hydrogen in the hydrogen bond acceptor’s position, it is much more favorable to allow the hydrogen bond to form.[9]
Solvents
Solvents also have an effect on allylic strain. When used in conjunction with knowledge of the effects of polarity on allylic strain, solvents can be very useful in directing the conformation of a product that contains an allylic structure in its transition state. When a bulky and polar solvent is able to interact with one of the substituents in the allylic group, the complex of the solvent can energetically force the bulky complex out of the allylic strain in favor of a smaller group.[10]
Conjugation
Conjugation increases the allylic strain because it forces substituents into a configuration that causes their atoms to be in closer proximity, increasing the strength of repulsive Van der Waals forces.[11] This situation occurs most noticeably when carboxylic acid or ketone is involved as a substituent of the allylic group. Resonance effect on the carboxylic group shifts the CO double bond to a hydroxy group. The carboxylic group will thus function as a hydroxyl group that will cause a large allylic strain to form and cancel the stabilization effects of the extended conjugation. This is very common in enolization reactions[2] and can be viewed in the figure below under "Acidic Conditions."
In situations where the molecule can either be in a conjugated system or avoid allylic strain, it has been shown that the molecule's major form will be the one that avoids strain. This has been found via the cyclization in the figure below.[12] Under treatment of perchloric acid, molecule A cyclizes into the conjugated system show in molecule B. However, the molecule will rearrange (due to allylic strain) into molecule C, causing molecule C to be the major species. Thus, the magnitude of destabilization via the allyic strain outweighs the stabilization caused by the conjugated system.[2]
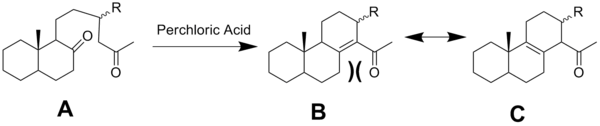
Acidic conditions
In cases where an enolization is occurring around an allylic group (usually as part of a cyclic system), A1,3 strain can cause the reaction to be nearly impossible. In these situations, acid treatment would normally cause the alkene to become protonated, moving the double bond to the carboxylic group, changing it to a hydroxy group. The resulting allylic strain between the alcohol and the other group involved in the allylic system is so great that the reaction can not occur under normal thermodynamic conditions.[13] This same enolization occurs much more rapidly under basic conditions, as the carboxylic group is retained in the transition state and allows the molecule to adopt a conformation that does not cause allylic strain.[13]
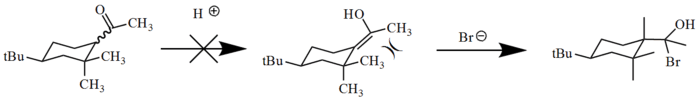
Application of allylic strain in organic reactions and total synthesis
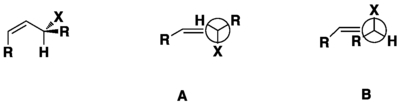
Origin of stereoselectivity of organic reactions from allylic strain
When one is considering allylic strain, one needs to consider the possible conformers and the possible stereoelectronic demand of the reaction. For example, in the conformation of (Z)-4-methylpent-2-ene, the molecule isn't frozen in the favored conformer but rotates in the dihedral angle around 30° at <1kcal/mol cost. In stereoselective reactions, there are 2 effects of allylic strain on the reaction which is the sterics effect and the electronic effects. The sterics effect is where the largest group prefer to be the farthest from the alkene. The electronic effect is where the orbitals of the subsituents prefer to align anti or outside of the orbitals depending on the reaction.[14]
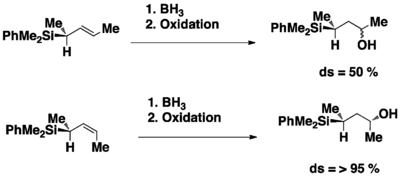
Hydroboration reaction
The hydroboration reaction is a useful reaction to functionalize alkenes to alcohols. In the reaction the trimethylsilyl (TMS) group fulfill 2 roles in directing the stereoselectivity of the reaction. First, the bulky size of TMS helped the molecule to preferably adopt a conformation where the TMS is not close to the methyl group on the alkene. Second, the TMS group conferred a stereoelectronic effect on the molecule by adopting an anti conformation to the directing orbitals of the alkene. For the regioselectivity of the reaction, the TMS group can stabilize the developing partial positive charge on the secondary carbon a lot better than a methyl group.[15]
Aldol reaction
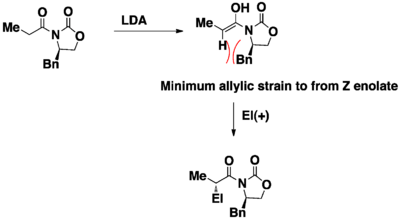
In the highly versatile and widely used Evans’ Aldol Reaction,[16] allylic strain played a major role in the development of the reaction. The Z enolate was created to avoid the allylic strain with oxazolidinone. The formation of a specific enolate enforces the development of relative stereochemistry throughout the reaction, making the aldol reaction a very predictive and useful methodology out there to synthesize chiral molecules. The absolute stereochemistry is then determined by the chirality of the oxazolidinone.
There is another aspect of aldol reaction that is influenced by the allylic strain. On the second aldol reaction, the product which is a 1,3 dicarbonyl is formed in high diastereoselectivity. This is because the acidity of the proton is significantly reduced because for the deprotonation to occur, it will have to go through a developing allylic strain in the unfavored conformation. In the favored conformation, the proton is not aligned properly for deprotonation to occur.

Diels-Alder reaction
In an intramolecular Diels-Alder reaction, asymmetric induction can be induced through allylic 1,3 strain on the diene or the dienophile. In the following example,[17] the methyl group on the dienophile forced the molecule to adopt that specific 6-membered ring conformation on the molecule.

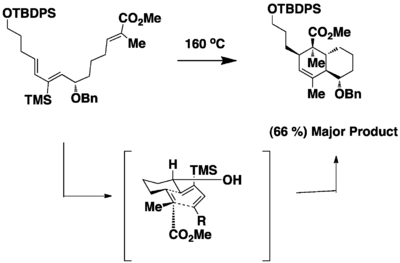
In the model studies to synthesize chlorothricolide,[18] an intramolecular Diels Alder reaction gave a mixture of diastereomers. But by installing the a bulky TMS substituent, the reaction gave the desired product in high diastereoselectivity and regioselectivity in good yield. The bulky TMS substituent helps enhance allylic 1,3 strain in the conformation of the molecule.
Total synthesis of natural products
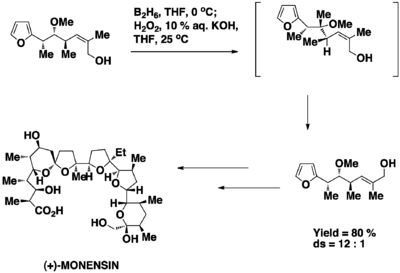
In the seminar paper on the total synthesis of (+)-monensin,[19] Kishi and co-workers utilized the allylic strain to induce asymmetric induction in the hydroboration oxidation reaction. The reaction is regioselective and stereoselective. The regioselectivity of the reaction is due to the significant positive character developed at the tertiary carbon. The stereoselectivity of the reaction is due to the attack by the borane from the least hindered side to which is where the methyl group lies at.
References
- Eric V. Anslyn and Dennis A. Dougherty Modern Physical Organic Chemistry University Science Books, 2006.
- Johnson, F (1968). "Allylic Strain in Six-Membered Rings". Chem. Rev. 68 (4): 375–413. doi:10.1021/cr60254a001.
- Johnson, F; Malhorta, S. K (1965). "Steric Interference in Allylic and Pseudo-Allylic Systems. I. Two Stereochemical Theorems". J. Am. Chem. Soc. 87 (23): 5492–5493. doi:10.1021/ja00951a047.
- Allinger, N. L.; Hirsch, Jerry A.; Miller, Mary Ann.; Tyminski, Irene J. (1968). "Conformational Analysis. LXIV. Calculation of the Structures and Energies of Unsaturated Hydrocarbons by the Westheimer Method". J. Am. Chem. Soc. 90 (21): 5773–5780. doi:10.1021/ja01023a021.
- Eliel, E. L.; Allinger, N. L.; Angyal, S. J.; Morrison, G. A. Conformational Analysis Interscience Publishers, Inc., New York, N. Y., 1965.
- Brown, H.; Barbarahs, G. K.; Berneis, H. L.; Bonner, W. H.; Johannesen, M. G.; Grayson, M. (1953). "Strained Homomorphs. 14. General Summary". J. Am. Chem. Soc. 75 (1): 1–6. doi:10.1021/ja01097a001.
- Hoffman, R (1989). "Allylic 1,3-strain as a controlling factor in stereoselective transformations". Chem. Rev. 89 (8): 1841–1860. doi:10.1021/cr00098a009.
- Bach, T; Jodicke K; Kather, K; Frohlich, R (1997). "1,3-Allylic Strain as a Control Element in the Paterno-Buchi Reaction of Chiral Silyl Enol Ethers: Synthesis of Diastereomerically Pure Oxetanes Containing Four Contiguous Stereogenic Centers". J. Am. Chem. Soc. 119 (10): 5315–5316. doi:10.1021/ja963827v.
- Ramey, B.; Gardner, P (1967). "Mechanism of photochemical alcohol addition to alpha, beta-unsaturated ketones". J. Am. Chem. Soc. 89 (15): 3949–3950. doi:10.1021/ja00991a078.
- McGarvey, G; Williams, J (1985). "Stereoelectronic controlling features of allylic asymmetry. Application to ester enolate alkylations". J. Am. Chem. Soc. 107 (5): 1435–1437. doi:10.1021/ja00291a067.
- Harris, R. K.; Sheppard, N. (1967). "Comments on the ring inversion of cyclohexane studied by NMR". J. Mol. Spectrosc. 23 (2): 231–235. Bibcode:1967JMoSp..23..231H. doi:10.1016/0022-2852(67)90015-X.
- Overton, K. H.; Renfrew, A. J. (1967). "The configuration at C-13 in labdanolic and eperuic acids". J. Chem. Soc. C: 931–935. doi:10.1039/J39670000931.
- Vaughn, W. R.; Caple, R; Csapilla, J; Scheiner, P (1965). "β-Bromo Acids. II. Solvolysis of Cyclic β-Bromo Acids". J. Am. Chem. Soc. 87 (10): 2204. doi:10.1021/ja01088a020.
- Houk K. N.; Paddon-Row, M.; Rondan, N.; Wu, Y.; Brown, F.; Spellmeyer, D.; Metz, J.; Li, Y; Loncharich, R.; et al. (1986). "Theory and Modeling of Stereoselective Organic Reactions". Science. 231 (4742): 1108. Bibcode:1986Sci...231.1108H. doi:10.1126/science.3945819.
- Fleming, I. (1988). "Stereocontrol in organic synthesis using silicon compounds". Pure Appl. Chem. 60: 71–78. doi:10.1351/pac198860010071.
- Evans, D. A.; Takacs, J. M.; McGee, L. R.; Ennis, M. D.; Mathre, D. J.; Bartroli, J.; et al. (1981). "Chiral enolate design". Pure Appl. Chem. 53 (6): 1109. doi:10.1351/pac198153061109.
- Ichihara, A.; et al. (1986). "Stereoselective total synthesis and stereochemistry of diplodiatoxin, a mycotoxin from ?". Tetrahedron Lett. 27 (12): 1347–1350. doi:10.1016/S0040-4039(00)84255-0.
- Roush, W. R.; Kageyama, Masanori; Riva, Renata; Brown, Bradley B.; Warmus, Joseph S.; Moriarty, Kevin J.; et al. (1991). "Enantioselective synthesis of the bottom half of chlorothricolide. 3. Studies of the steric directing group strategy for stereocontrol in intramolecular Diels-Alder reactions". J. Org. Chem. 56 (3): 1192. doi:10.1021/jo00003a049.
- Nicolaou, K. C.; et al. Classics in Total Synthesis. Wiley. p. 185.
| Wikimedia Commons has media related to Allylic strain. |
External links
- Advanced Organic Chemistry Lecture Notes (Evans, D. A.; Myers, A. G. Harvard University, 2006-2007)