Phakomatosis
Phakomatoses, or phacomatosis pigmentovascularis (PPV),[1] is the term used for a group of rare syndromes[2] involving structures arising from the embryonic ectoderm. These are characterised by vascular and pigmentary birthmarks[1] or skin lesions, and often involving multiple organ systems in the body.[2] The term is used to describe the “association of a vascular nevus with an extensive pigmentary nevus”.[3]
| Phakomatoses | |
|---|---|
| Other names | Phakomatosis, PPV, Phacomatosis pigmentovascularis |
| Specialty | Medicine, Dermatology |
| Symptoms | Dermal, ocular and tumour expressions |
| Complications | Systemic involvement |
| Usual onset | Genetic predisposition, with more research required |
| Duration | Lifelong |
| Causes | Genetic causes |
| Treatment | Some expressions require no treatment, while others require lifelong monitoring of tumours, laser therapy, or removal of tumours or growths. |
These multi-system disorders involve the ectodermal structures like central nervous system, skin and eyes.[4] The lesions have a variable severity.[5][6] However, it has been subsequently found that mesodermal and endodermal tissues too are involved.
These diseases often involve a disorder of non-cancerous skin cells,[2] melanocytes, which produce pigment appearing as moles, birthmarks[2] or growths[3] on the surface of the skin or just beneath,[2] which can be rough, flat or raised.[2] These dermal growths, including port wine stains, moles, discolouration, café au lait spots and hyperpigmentation, are often composed of blood vessels[7] which can cause functional or cosmetic problems,[2] as well as benign tumours located on the surface or just below of the skin.[2]
These physical symptoms are caused by systemic tumour-like growths of the eye, brain, skin, internal organs and bones.[3] The tendency of the condition to involve multiple bodily systems means the conditions are also known as ocular-neurocutaneous syndromes,[3] with about half of people with PPV[1] experiencing systemic symptoms, often suffering from neurological, ocular or muscular abnormalities.[1]
These conditions are regularly present at birth, though there are instances of the syndromes appearing later on in life.[3]
History
The term ‘phakomatosis’ originated in 1923, when Dutch ophthalmologist van der Hoeve,[3] used the term “phakoma” to refer to a “mother spot or birthmark”,[3] a physical characteristic common to many of the confirmed cases.[3] The term ‘phakomatoses’ was derived from “phakos”,[8] the Greek term for birthmark.[8] He originally used the phrase to describe two diseases: neurofibromatosis and tuberous sclerosis.[8]
The first documented cases of phakomatoses are said to date back to 1910.[3]
It has since become a spectrum of diseases, with other neurocutaneous diseases being incorporated under the umbrella term.[8]
In 1985, phakomatoses became the term used to describe four types of conditions, differentiated by the pigmented lesions associated with each of them.[3]
Since the term became recognised in the medical world, most reports of PPV have been from Japan, Mexico and Argentina.[3] The most commonly presented condition is neurofibromatosis Type IIb (45% of cases),[1] followed by neurofibromatosis type IIa (30% of cases).[1] The remaining types of phakomatoses have had far less reported cases.[3]
Now, the term is used to refer to several primary conditions, all within the ‘phakomatoses’ umbrella, including:
- Von Recklinghausen syndrome (neurofibromatosis) – Type I and Type II
- Bourneville syndrome (tuberous sclerosis)
- Sturge-Weber syndrome (encephalofacial hemangiomatosis)
- Von Hippel-Lindau syndrome (hemangiomatosis)
Types of Phakomatoses: Signs and Characteristics
Neurofibromatosis Type I (von Recklinghausen disease)
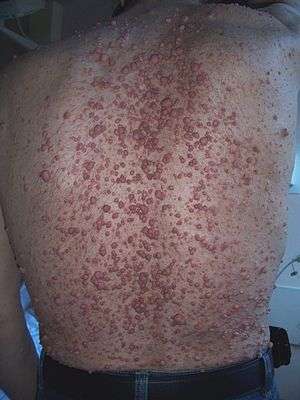
Neurofibromatosis Type I is a genetic disorder, caused by the affected individual inheriting one copy of a mutant gene and one normal gene on a pair of autosomal chromosomes.[9] The condition results in multiple neurofibromas[9] (benign nerve-sheath tumours in the peripheral nervous system),[8] and occasionally neurilemmomas.[9]
Neurofibromas are often asymptomatic, unless they exert pressure on areas of the body, such as the spinal chord nerve roots.[9] These tumours may disfigure the appearance of affected areas, as does skin hyperpigmentation, which is common in this condition.[9]
Non-tumour characteristics of Neurofibromatosis Type I include café-au-lait spots, skin fold freckling,[8] Iris Lisch nodules and optic glycoma in the eye, and skeletal displasias.[8]
Less common complications include epilepsy and learning impairments,[9] with studies finding at least half of children with Neurofibromatosis Type I suffer from learning difficulties, many with attention deficit disorder.[8] In rare cases, Neurofibromatosis Type I has been linked to neuroendocrine adenoma syndrome,[9] and has contributed to early mortality owing to malignant tumours and vascular disease.[8]
Neurofibromatosis Type II
Neurofibromatosis Type II is characterised by bilateral vestibular schwannomas,[8] with affected individuals also often experiencing schwannomas of other cranial nerves, spinal nerve roots and peripheral nerves. These schwannomas can occur within the skin, appearing as a thickening patch with hair growth. The syndrome can also cause tumours within the nervous system, such as meningiomas, ependymomas, gliomas, and neurofibromas.[8] Unlike Neurofibromatosis Type I, café-au-lait spots[8] and skin lesions common to type I[9] are rarely seen in Type II.
Non-tumour manifestations can include cataracts and deafness.[9]
Tuberous sclerosis (Bourneville syndrome)

Another branch of phakomatosis, Tuberous sclerosis is also caused by an autosomal dominant trait,[9] and is characterised by leaf-shaped depigmented moles on the skin, epilepsy, learning impairments, and angiofibromas, or dome-shaped lesions normally found on the cheeks, nose, forehead and chin.[9] The facial tumours regularly identify the condition.[9]
This condition affects multiple bodily systems, including the brain, skin, heart, eye, kidneys and other tissues.[8] Most commonly, the syndrome causes neurological complications, especially epilepsy and seizures[9],,[8] learning difficulties[9],,[8] diabetes mellitus, and other cardiac and renal complications.[9] Dental defects including hyperplastic gingivitis are also symptomatic of this condition.[9]
Sturge-Weber Syndrome (encephalotrigeminal angiomatosis)
Sturge-Weber syndrome is most commonly characterised by a red or pink “port-wine” birthmark, and an angioma, often located on the upper face or skull.[9] The condition interferes with the development of blood vessels in the body, leading to complications in the brain, skin and eyes from birth. Individuals with the syndrome also often present with a brain abnormality,[1] leptomeningeal angioma, or glaucoma in the eye.[3] These three physical symptoms can occur in varying degrees of severity, and many individuals with the syndrome won’t present all three complications.[3]
The impact of this condition on the brain is dependent on the location of the angioma. If located on the lower face, the parietal and frontal lobes of the brain may be impacted,[9] in addition to the occipital lobe of the brain, which is commonly affected.[9] Other complications arising from this condition include convulsions, partial paralysis of the body, and learning difficulties.[9]
Von Hippel-Lindau syndrome (hemangiomatosis)
Von Hippel-Lindau syndrome is most commonly identified by the occurrence of hemangioblastomas, tumours made up of vascular elements,[8] particularly in the cerebellum, retina, and spinal cord.[8] Endolymphatic sac tumours are also common[8] and can lead to deafness, as are renal cysts, and cysts in the pancreas, liver and epididymis.[8]
Genetic Causes
There is some debate surrounding the genetic causes of the phakomatoses, with certain types found to be caused by genetic factors, and others not[10],.[11]
Studies have found that some cases of phakomatosis are caused by a somatic mutation in the GNA11 or GNAQ gene, which is present only in affected tissues within the body.[1] As these mutations are not found in the blood or unaffected areas of the body, this suggests the syndromes can be caused by non-inherited mutations, randomly acquired after conception.[1] No evidence of hereditary cause has been discovered in Sturge-Weber Syndrome.[10]
However, some types of phakomatoses have been found to be caused by genetic factors.[10] Clinical studies have found Von Hippel-Lindau syndrome to be caused by a dominant inherited or de novo germline mutation,[10] with the inherited familial cancer syndrome predisposing individuals for genetic mutations in the VHL gene.[10] In fact, in a clinical study, 80% of individuals with Von Hippel-Lindau syndrome had an affected parent.[10]
Similarly, while individuals presenting with Neurofibromatosis Type I, cases where both parents were unaffected were extremely rare, suggesting some genetic correlation,[8] though more research is required to determine the genetic causes.[8] Unlike Type I, research suggests a genotype-phenotype correlation in individuals diagnosed with Neurofibromatosis Type II,[8] with individuals with whole gene deletions often experiencing milder manifestations of the condition.[8]
Further research is being done to determine the genetic or other causes of the remaining branches of phakomatoses.[11]
Diagnosis
As the phakomatoses are rare conditions, with a wide range of potential symptoms and complications in their clinical expression,[1] diagnosis can be complex. Indeed, most research and literature only assess isolated cases[1] for commonalities.
Clinical research conducted by Fernández-Guarino, M., Boixeda, P., de las Heras, E., Aboin, S., García-Millán, C. and Olasolo, P. in 2008, analysing 15 cases of phakomatoses found that the diagnosis of these syndromes is mostly clinical.[1]
The most common clinical expression of phakomatoses is the presence of nevus anemicus, or paler patches of skin, which was present in 50% of cases considered in Fernández-Guarino et. al.’s research.[1] Café-au-lait spots and depigmentation were also found to be common distinguishing characteristics in diagnosis, though were not present in all cases.[1]
Fernández-Guarino et. al.’s clinical trial found that an estimated 50% of patients with phakomatoses are subject to systemic symptoms.[2] It was found that neurological conditions developed in the first few months of life, if at all.[2]
The abnormalities caused by the phakomatoses are difficult to determine, as the research has found significantly varied abnormalities present in different patients,[2] and resulting from the different syndromes. The most common ocular abnormality has been proven to be ocular melanosis,[2] a congenital eye disease. New clinical findings continue to emerge surrounding potential abnormalities[12] as a result of these conditions.
Diagnosis
While most cases of phakomatoses, particularly those without systematic complications, are benign and require no treatment,[3] some cases simply require the removal of the resulting mole, dermal lesion or raised tumour.[3] Treatment of Von Hippel-Lindau syndrome is simply surgical removal,[8] with individuals diagnosed with VHL requiring lifelong observation[8] for potential tumours.
The instances without systemic complications, particularly Sturge-Weber syndrome, only require treatment to prevent impacts to affected individuals’ body image and self-esteem,[2] particularly given lesions may grow as the body grows[2],.[12] Treatment and removal of these lesions depends on the individual and the symptoms experienced, however it is suggested that the earlier the removal of dermal expressions, the better.[2]
Research by Fernández-Guarino et. al. found that treatment of port-wine stains using a pulsed dye laser,[2] and treatment of the skin marks/moles using Q-switched lasers,[3] may enhance the individual’s quality of life.
Clinical trials are underway, with some success to date, to discover a more reliable approach to treatment.[3]
References
- "Phacomatosis pigmentovascularis | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2020-05-25.
- Shields, Carol L.; Shields, Jerry A. (2013), "Phakomatoses", Retina, Elsevier, pp. 2170–2183, doi:10.1016/b978-1-4557-0737-9.00132-6, ISBN 978-1-4557-0737-9
- Fernández-Guarino, Montse; Boixeda, Pablo; de las Heras, Elena; Aboin, Sonsoles; García-Millán, Cristina; Olasolo, Pedro Jaén (January 2008). "Phakomatosis pigmentovascularis: Clinical findings in 15 patients and review of the literature". Journal of the American Academy of Dermatology. 58 (1): 88–93. doi:10.1016/j.jaad.2007.08.012. PMID 18045734.
- Neau, JP; Godeneche, G; Mathis, S; Guillet, G (2014). "Neurodermatology". Handbook of Clinical Neurology. 121: 1561–94. doi:10.1016/B978-0-7020-4088-7.00104-8. ISBN 9780702040887. PMID 24365436.
- Arthur Rook; Tony Burns (FRCP.) (2004). Rook's textbook of dermatology. Wiley-Blackwell. pp. 5–. ISBN 978-0-632-06429-8. Retrieved 27 October 2010.
- Barbagallo, JS; Kolodzieh, MS; Silverberg, NB; Weinberg, JM (Jul 2002). "Neurocutaneous disorders". Dermatologic Clinics. 20 (3): 547–60, viii. doi:10.1016/s0733-8635(02)00005-0. PMID 12170887.
- Shields, Carol L.; Shields, Jerry A. (2013), "Phakomatoses", Retina, Elsevier, pp. 2170–2183, doi:10.1016/b978-1-4557-0737-9.00132-6, ISBN 978-1-4557-0737-9
- Huson, Susan M.; Korf, Bruce R. (2013), "The Phakomatoses", Emery and Rimoin's Principles and Practice of Medical Genetics, Elsevier, pp. 1–45, doi:10.1016/b978-0-12-383834-6.00128-2, ISBN 978-0-12-383834-6
- Scully, Crispian (2014), Scully's Medical Problems in Dentistry, Elsevier, pp. 3–23, doi:10.1016/b978-0-7020-5401-3.00001-1, ISBN 978-0-7020-5401-3 Missing or empty
|title=(help);|chapter=ignored (help) - Perlman, Susan (2018), Neurogenetics, Part II, Handbook of Clinical Neurology, Elsevier, 148, pp. 823–826, doi:10.1016/b978-0-444-64076-5.00053-3, ISBN 978-0-444-64076-5, PMID 29478617 Missing or empty
|title=(help);|chapter=ignored (help) - Ruiz-Maldonado, Ramón; Tamayo, Lourdes; Laterza, Amelia M.; Brawn, Gabriela; Lopez, Arturo (November 1987). "Phacomatosis Pigmentovascularis: A New Syndrome? Report of Four Cases". Pediatric Dermatology. 4 (3): 189–196. doi:10.1111/j.1525-1470.1987.tb00777.x. ISSN 0736-8046. PMID 3422849.
- Iyer, Ramesh S. (29 September 2015). Pediatric imaging : the essentials. Chapman, Teresa, 1972-. Philadelphia. ISBN 978-1-4963-2956-1. OCLC 938870724.