Tsuji–Trost reaction
The Tsuji–Trost reaction (also called the Trost allylic alkylation or allylic alkylation) is a palladium-catalysed substitution reaction involving a substrate that contains a leaving group in an allylic position. The palladium catalyst first coordinates with the allyl group and then undergoes oxidative addition, forming the π-allyl complex. This allyl complex can then be attacked by a nucleophile, resulting in the substituted product.[1]
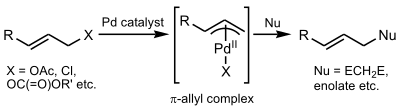
This work was first pioneered by Jiro Tsuji in 1965[2] and, later, adapted by Barry Trost in 1973 with the introduction of phosphine ligands.[3] The scope of this reaction has been expanded to many different carbon, nitrogen, and oxygen-based nucleophiles, many different leaving groups, many different phosphorus, nitrogen, and sulfur-based ligands, and many different metals (although palladium is still preferred).[4] The introduction of phosphine ligands led to improved reactivity and numerous asymmetric allylic alkylation strategies. Many of these strategies are driven by the advent of chiral ligands, which are often able to provide high enantioselectivity and high diastereoselectivity under mild conditions. This modification greatly expands the utility of this reaction for many different synthetic applications. The ability to form carbon-carbon, carbon-nitrogen, and carbon-oxygen bonds under these conditions, makes this reaction very appealing to the fields of both medicinal chemistry and natural product synthesis.
History
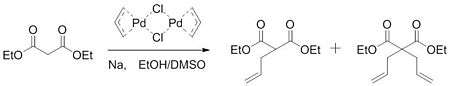
In 1962, Smidt published work on the palladium-catalysed oxidation of alkenes to carbonyl groups. In this work, it was determined that the palladium catalyst activated the alkene for the nucleophilic attack of hydroxide.[5] Gaining insight from this work, Tsuji hypothesized that a similar activation could take place to form carbon-carbon bonds. In 1965, Tsuji reported work that confirmed his hypothesis. By reacting an allylpalladium chloride dimer with the sodium salt of diethyl malonate, the group was able to form a mixture of monoalkylated and dialkylated product.[6]
The scope of the reaction was expanded only gradually until Trost discovered the next big breakthrough in 1973. While attempting to synthesize acyclic sesquiterpene homologs, Trost ran into problems with the initial procedure and was not able to alkylate his substrates. These problems were overcome with the addition of triphenylphosphine to the reaction mixture.
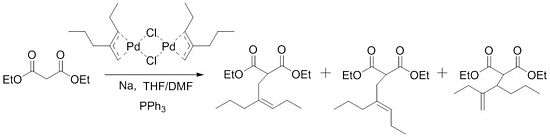
These conditions were then tested out for other substrates and some led to "essentially instantaneous reaction at room temperature." Soon after, he developed a way to use these ligands for asymmetric synthesis.[7] Not surprisingly, this spurred on many other investigations of this reaction and has led to the important role that this reaction now holds in synthetic chemistry.
Mechanism
Starting with a zerovalent palladium species and a substrate containing a leaving group in the allylic position, the Tsuji–Trost reaction proceeds through the catalytic cycle outlined below.
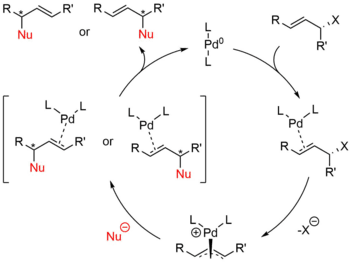
First, the palladium coordinates to the alkene, forming a η2 π-allyl-Pd0 Π complex. The next step is oxidative addition in which the leaving group is expelled with inversion of configuration and a η3 π-allyl-PdII is created (also called ionization). The nucleophile then adds to the allyl group regenerating the η2 π-allyl-Pd0 complex. At the completion of the reaction, the palladium detaches from the alkene and can start again in the catalytic cycle.[8]
"Hard" versus "soft" nucleophiles
The nucleophiles used are typically generated from precursors (pronucleophiles) in situ after their deprotonation with base.[9] These nucleophiles are then subdivided into "hard" and "soft" nucleophiles using a paradigm for describing nucleophiles that largely rests on the pKas of their conjugate acids. "Hard" nucleophiles typically have conjugate acids with pKas greater than 25, while "soft" nucleophiles typically have conjugate acids with pKas less than 25.[10] This descriptor is important because of the impact these nucleophiles have on the stereoselectivity of the product. Stabilized or "soft" nucleophiles invert the stereochemistry of the π-allyl complex. This inversion in conjunction with the inversion in stereochemistry associated with the oxidative addition of palladium yields a net retention of stereochemistry. Unstabilized or "hard" nucleophiles, on the other hand, retain the stereochemistry of the π-allyl complex, resulting in a net inversion of stereochemistry.[11]
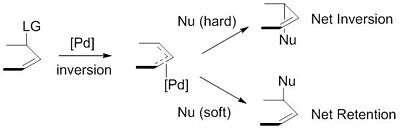
This trend is explained by examining the mechanisms of nucleophilic attack. "Soft" nucleophiles attack the carbon of the allyl group, while "hard" nucleophiles attack the metal center, followed by reductive elimination.[12]
Phosphine ligands
Phosphine ligands, such as triphenylphosphine or the Trost ligand, have been used to greatly expand the scope of the Tsuji–Trost reaction. These ligands can modulate the properties of the palladium catalyst such as steric bulk as well as the electronic properties. Importantly, these ligands can also instill chirality to the final product, making it possible for these reactions to be carried out asymmetrically as shown below.
Allylic asymmetric substitution
The enantioselective version of the Tsuji–Trost reaction is called the Trost asymmetric allylic alkylation (Trost AAA) or simply, asymmetric allylic alkylation (AAA). These reactions are often used in asymmetric synthesis.[13][14][15] The reaction was originally developed with a palladium catalyst supported by the Trost ligand, although suitable conditions have greatly expanded since then. Enantioselectivity can be imparted to the reaction during any of the steps aside from the decomplexation of the palladium from the alkene since the stereocenter is already set at that point. Five main ways have been conceptualized to take advantage of these steps and yield enantioselective reaction conditions. These methods of enantiodiscrimination were previously reviewed by Trost:
- Preferential Ionization via Enantioselective Olefin Complexation
- Enantiotopic Ionization of Leaving Groups
- Attack at Enantiotopic Termini of the Allyl Complex
- Enantioface Exchange in the π-Allyl Complex
- Differentiation of Prochiral Nucleophile Faces
The favored method for enantiodiscrimination is largely dependent on the substrate of interest, and in some cases, the enantioselectivity may be influenced by several of these factors.
Scope
Nucleophiles
Many different nucleophiles have been reported to be effective for this reaction. Some of the most common nucleophiles include malonates, enolates, primary alkoxides, carboxylates, phenoxides, amines, azides, sulfonamides, imides, and sulfones.
Leaving groups
The scope of leaving groups has also been expanded to include a number of different leaving groups, although carbonates, phenols, phosphates, halides and carboxylates are the most widely used.
"Hard" and "soft" nucleophiles
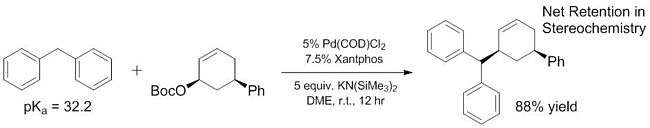
Recent work has demonstrated that the scope of "soft" nucleophiles can be expanded to include some pronucleophiles that have much higher pKas than ~ 25. Some of these "soft" nucleophiles have pKas ranging all the way to 32,[16] and even more basic pronucleophiles (~44) have been shown to act as soft nucleophiles with the addition of Lewis acids that help to facilitate deprotonation.[17] The improved pKa range of "soft" nucleophiles is critical because these nucleophiles are the only ones that have been explored[18][19] for enantioselective reactions until very recently[20] (although non-enantioselective reactions of "hard" nucleophiles have been known for some time[21]). By increasing the scope of pronucleophiles that act as "soft" nucleophiles, these substrates can also be incorporated into enantioselective reactions using previously reported and well characterized methods.
Ligands
Building on the reactivity of the triphenylphosphine ligand, the structure of ligands used for the Tsuji–Trost reaction quickly became more complex. Today, these ligands may contain phosphorus, sulfur, nitrogen or some combination of these elements, but most studies have concentrated on the mono- and diphosphine ligands. These ligands can be further classified based on the nature of their chirality, with some ligands containing central chirality on the phosphorus or carbon atoms, some containing biaryl axial chirality, and others containing planar chirality. Diphosphine ligands with central chirality emerged as an effective type of ligand (particularly for asymmetric allylic alkylation procedures) with the Trost Ligand being one such example.[22] Phosphinooxazolines (PHOX) ligands have been employed in the AAA, particularly with carbon-based nucleophiles.[23]
Additional substrates
The reaction substrate has also been extended to allenes. In this specific ring expansion the AAA reaction is also accompanied by a Wagner–Meerwein rearrangement:[24][25]

Applications
Pharmaceutical/natural products synthesis
The ability to form carbon-carbon, carbon-nitrogen, and carbon-oxygen bonds enantioselectively under mild conditions makes the Trost asymmetric allylic alkylation extremely appealing for the synthesis of complex molecules.
An example of this reaction is the synthesis of an intermediate in the combined total synthesis of galantamine and morphine[26] with 1 mol% [pi-allylpalladium chloride dimer], 3 mol% (S,S) Trost ligand, and triethylamine in dichloromethane at room temperature. These conditions result in the formation of the (−)-enantiomer of the aryl ether in 72% chemical yield and 88% enantiomeric excess.

Another Tsuji–Trost reaction was used during the initial stages of the synthesis of (−)-Neothiobinupharidine. This recent work demonstrates the ability of this reaction to give highly diastereoselective (10:1) and enantioselective (97.5:2.5) products from achiral starting material with only a small amount of catalyst (1%).[27]
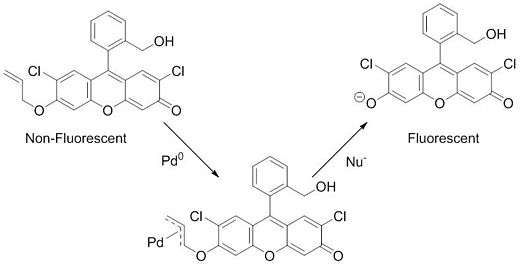
Palladium detection
Aside from the practical application of this reaction in medicinal chemistry and natural product synthesis, recent work has also used the Tsuji–Trost reaction to detect palladium in various systems. This detection system is based on a non-fluorescent fluorescein-derived sensor (longer-wavelength sensors have also recently been developed for other applications[28]) that becomes fluorescent only in the presence of palladium or platinum. This palladium/platinum sensing ability is driven by the Tsuji–Trost reaction. The sensor contains an allyl group with the fluorescein functioning as the leaving group. The π-allyl complex is formed and after a nucleophile attacks, the fluorescein is released, yielding a dramatic increase in fluorescence.[29][30]
This simple, high-throughput method to detect palladium by monitoring fluorescence has been shown to be useful in monitoring palladium levels in metal ores,[31] pharmaceutical products,[32] and even in living cells.[33] With the ever-increasing popularity of palladium catalysis, this type of quick detection should be very useful in reducing the contamination of pharmaceutical products and preventing the pollution of the environment with palladium and platinum.
External links
- Org. Synth. 1989, 67, 105
- Org. Synth. 2009, 86, 47
- example of tsuji-trost reaction in total synthesis see : http://www.biocis.u-psud.fr/IMG/pdf/concise_total_synthesis_of_Minfiensine.pdf the second reaction found on website of the biocis team : http://www.biocis.u-psud.fr/spip.php?article332
References
- Strategic Applications of Named Reactions in Organic Synthesis (Paperback) by Laszlo Kurti, Barbara Czako ISBN 0-12-429785-4
- Organic syntheses by means of noble metal compounds XVII. Reaction of π-allylpalladium chloride with nucleophiles Tetrahedron Letters, Volume 6, Issue 49, 1965, Pages 4387-4388 Jiro Tsuji, Hidetaka Takahashi, Masanobu Morikawa doi:10.1016/S0040-4039(00)71674-1
- Trost, B. M.; Fullerton, T. J. "New synthetic reactions. Allylic alkylation." J. Am. Chem. Soc. 1973, 95, 292–294. doi:10.1021/ja00782a080.
- Rios, Itzel Guerrero; Rosas-Hernandez, Alonso; Martin, Erika; "Recent Advances in the Application of Chiral Phosphine Ligands in Pd-Catalysed Asymmetric Allylic Alkylation." Molecules, 2011, 16 970–1010. doi:10.3390/molecules16010970
- Smidt, J., Hafner, W., Jira, R., Sieber, R., Sedlmeier, J. and Sabel, A. (1962), Olefinoxydation mit Palladiumchlorid-Katalysatoren. Angewandte Chemie, 74: 93–102. doi:10.1002/ange.19620740302
- Organic syntheses by means of noble metal compounds XVII. Reaction of π-allylpalladium chloride with nucleophiles Tetrahedron Letters, Volume 6, Issue 49, 1965, pages 4387–4388 Jiro Tsuji, Hidetaka Takahashi, Masanobu Morikawa doi:10.1016/S0040-4039(00)71674-1
- Asymmetric Transition Metal-Catalyzed Allylic Alkylations Barry M. Trost David L. Van Vranken Chem. Rev., 1996, 96 (1), pp 395–422 doi:10.1021/cr9409804
- Trost, Barry M.; Zhang, Ting; Sieber, Joshua D.; "Catalytic asymmetric allylic alkylation employing heteroatom nucleophiles: a powerful method for C-X bond formation." Chem. Sci. 2010, 1, 427–440.
- Strategic Applications of Named Reactions in Organic Synthesis (Paperback) by Laszlo Kurti, Barbara Czako ISBN 0-12-429785-4
- Trost, B. M.; Thaisrivongs, D. A. J. Am. Chem. Soc. 2008, 130, 14092
- Luparia, Marco; Oliverira, Maria Teresa; Audisio, Davide; Frebault, Frederic; Goddard, Richard; Maulide, Nuno; "Catalytic Asymmetric Diastereodivergent Deracemization." Angew. Chem. Int. Ed. 2011, 50, 12631–12635.
- B. M. Trost, T. R. Verhoeven, J. M. Fortunak, Tetrahedron Lett. 1979, 20, 2301 – 2304
- Trost, B. M.; Dietsch, T. J. "New synthetic reactions. Asymmetric induction in allylic alkylations." J. Am. Chem. Soc. 1973, 95, 8200–8201. doi:10.1021/ja00805a056.
- Trost, B. M.; Strege, P. E. "Asymmetric induction in catalytic allylic alkylation." J. Am. Chem. Soc. 1977, 99, 1649–1651. doi:10.1021/ja00447a064.
- Asymmetric Transition-Metal-Catalyzed Allylic Alkylations:Applications in Total Synthesis Trost, B. M.; Crawley, M. L. Chem. Rev.; (Review); 2003; 103(8); 2921–2944. doi:10.1021/cr020027w
- Sha, Sheng-Chun; Zhang, Jiadi; Carroll, Patrick J.; Walsh, Patrick J.; "Raising the pKa Limit of "Soft" Nucleophiles in Palladium-Catalyzed Allylic Substitutions: Application of Diarylmethane Pronucleophiles." JACS. 2013, 135, 17602–17609. doi: 10.1021/ja409511n
- Zhang, J.; Stanciu, C.; Wang, B.; Hussain, M. M.; Da, C.-S.; Carroll, P. J.; Dreher, S. D.; Walsh, P. J. Palladium-Catalyzed Allylic Substitution with (η6-Arene–CH2Z)Cr(CO)3-Based Nucleophiles, J. Am. Chem. Soc. 2011, 133, 20552.
- Trost, B. M.; Toste, F. D. J. Am. Chem. Soc. 1999, 121, 4545.
- Trost, B. M.; Machacek, M. R.; Aponick, A. Acc. Chem. Res. 2006, 39, 747.
- Li, Xiao-Hui; Zheng, Bao-Hui; Ding, Chang-Hua; Hou, Xue-Long; "Enantioselective Synthesis of 2,3-Disubstituted Indanones via Pd-Catalyzed Intramolecular Asymmetric Allylic Alkylation of Ketones." Org. Lett. ASAP. doi: 10.1021/ol402980v
- Castanet, Y.; Petit, F. Tetrahedron Lett. 1979, 20, 3221.
- Lu, Zhan; Ma, Shengming; "Metal-Catalyzed Enantioselective Allylation in Asymmetric Synthesis." Angew. Chem. Int. Ed. 2008, 47, 258–297. doi: 10.1002/anie.200605113
- Behenna, D. C.; Stoltz, B. M., Shengming; "The Enantioselective Tsuji Allylation." J. Am. Chem. Soc. 2004, 126, 15044–15045. doi: 10.1021/ja044812x
- Trost, B. M.; Xie, J. "Palladium-Catalyzed Asymmetric Ring Expansion of Allenylcyclobutanols: An Asymmetric Wagner–Meerwein Shift." J. Am. Chem. Soc. 2006, 128, 6044–6045. doi:10.1021/ja0602501.
- The co-catalysts are benzoic acid and triethylamine. Molecular sieves (MS) prevent hydrolysis.
- Trost, B. M.; Tang, W.; Toste, F. D. "Divergent Enantioselective Synthesis of (−)-Galantamine and (−)-Morphine." J. Am. Chem. Soc. 2005, 127, 14785–14803. doi:10.1021/ja054449+.
- Jansen, Daniel J.; Shenvi, Ryan A.; "Synthesis of (−)-Neothiobinupharidine." JACS. 2013, 135, 1209–1212. doi: 10.1021/ja310778t
- Wang, Zhifei; Zheng, Shuang; Cai, Jin; Wang, Peng; Feng, Jie; Yang, Xia; Zhang, Liming; Ji, Min; Wu, Fugen; He, Nongyue; Wan, Neng; "Fluorescent Artificial Enzyme-Linked Immunoassay System Based on Pd/C Nanocatalyst and Fluorescent Chemodosimeter." Anal. Chem. ASAP. doi: 10.1021/ac403001y
- Garner, Amanda L.; Koide, Kazunori; "Studies of a fluorogenic probe for palladium and platinum leading to a palladium-specific detection method." Chem. Commun. 2009, 86–88. doi:10.1039/b814197e
- Song, Fengling; Garner, Amanda L.; Koide, Kazunori; "A Highly Sensitive Fluorescent Sensor for Palladium Based on the Allylic Oxidative Insertion Mechanism." JACS. 2007, 129, 12354–12355. doi: 10.1021/ja073910q
- Williams, Jessica M.; Koide, Kazunori; "A High-Throughput Method To Detect Palladium in Ores." Ind. Eng. Chem. Res. 2013, 52, 8612–8615. doi: 10.1021/ie400959z
- Bu, Xiaodong; Koide, Kazunori; Carder, Evan J.; Welch, Christopher J.; "Rapid Analysis of Residual Palladium in Pharmaceutical Development Using a Catalysis-Based Fluorometric Method." Org. Process Res. Dev. 2013, 17, 108–113. doi: 10.1021/op3003008
- Zhu, Baocun; Gao, Chenchen; Zhao, Yunzhou; Liu, Caiyun; Li, Yamin; Wei, Qin; Ma, Zhenmin; Du, Bin; Zhang, Xiaoling; "A 4-hydroxynaphthalimide-derived ratiometric fluorescent chemodosimeter for imaging palladium in living cells." Chem. Commun. 2011, 47, 8656–8658. doi: 10.1039/c1cc13215f