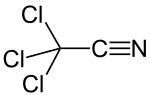
Trichloroacetonitrile
Trichloroacetonitrile is an organic compound with the formula CCl3CN. It is a colourless liquid, although commercial samples often are brownish. It is used commercially as a precursor to the fungicide etridiazole. It is prepared by dehydration of trichloroacetamide.[1] As a bifunctional compound, trichloroacetonitrile can react at both the trichloromethyl and the nitrile group. The electron withdrawing effect of the trichloromethyl group activates the nitrile group for nucleophilic additions. The high reactivity makes trichloroacetonitrile a versatile reagent, but also causes its susceptibility towards hydrolysis.
 | |
| Names | |
|---|---|
| IUPAC name
Trichloroacetonitrile | |
| Identifiers | |
3D model (JSmol) |
|
| ChemSpider | |
| ECHA InfoCard | 100.008.078 |
PubChem CID |
|
| UNII | |
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| C2Cl3N | |
| Molar mass | 144.38 g·mol−1 |
| Appearance | colourless liquid |
| Density | 1.44 g/mL |
| Melting point | -42 |
| Boiling point | 83 to 84 °C (181 to 183 °F; 356 to 357 K) |
| insoluble | |
| Hazards | |
| Main hazards | GHS06, GHS09 |
| Safety data sheet | MSDS |
| NFPA 704 (fire diamond) | |
| Flash point | 195 °C (383 °F; 468 K) |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |

Synthesis
The production of trichloroacetonitrile by dehydration of trichloroacetamide was first described in 1873 by L. Bisschopinck at the Katholieke Universiteit Leuven.[2]
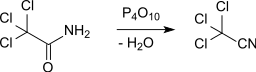
Trichloroacetonitrile can be obtained by chlorination of acetonitrile on a Zn, Cu and alkaline earth metal halides impregnated activated carbon catalyst at 200-400 °C in 54% yield.[3]
The high temperatures required by this process favours the formation of by-products, such as tetrachloromethane. In contrast, the chlorination of acetonitrile saturated with hydrogen chloride leads to pure trichloroacetonitrile even at 50-80 °C in good yields.[4]
Like other halogenated acetonitriles, trichloroacetonitrile is produced from organic substances such as algae, humic acids and proteinaceous material in the disinfecting chlorination of water from natural sources.[5][6]
Properties
Freshly distilled trichloroacetonitrile is a colorless, liquid with a pungent odor that discolours rapidly yellowish to light brown. It is sensitive towards water, acids and bases.
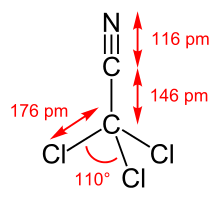
The bond lengths are 146.0 pm (C-C), 116.5 pm (C-N) and 176.3 pm (C-Cl). The bond angle is 110.0 ° (ClCCl).[7]
Use
The substitution of all electronegative substituents in trichloroacetonitrile by nucleophilic attack of alkoxide anions produces orthocarbonic acid esters in high yield.
Due to the high reactivity of the chlorine atoms, trichloroacetonitrile can be used (especially in combination with triphenylphosphine) to convert allylic alcohols into the corresponding allylic chlorides.[8]

With carboxylic acids, acyl chlorides are obtained.[9]
Due to the mild reaction conditions, the Cl3CCN/PPh3 system is also suitable for the activation of carboxylic acids and their linkage with supported amino compounds to amides (peptides) in solid-phase syntheses.[10] From sulfonic acids, the corresponding sulfochlorides are formed analogously.[11] In an analogous manner, the activation of diphenylphosphoric acid with Cl3CCN/PPh3 and reaction with alcohols or amines proceeds to the corresponding phosphoric acid esters or amides in a gentle and efficient one-pot reaction.[12]
Also, phenolic hydroxy groups in nitrogen-containing aromatics can be converted into the chlorine compounds.[13]
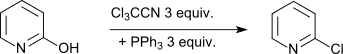
In a Hoesch reaction, aromatic hydroxyketones are formed in the reaction of substituted phenols with trichloroacetonitrile, for example from 2-methyl phenol the 2-trichloroacyl derivative in 70% yield.[14]

The electron-withdrawing effect of the trichloromethyl group activates the nitrile group of trichloroacetonitrile for the attack of nucleophilic oxygen, nitrogen and sulfuric compounds. For example, alcohols give under base catalysis in a direct and reversible addition O-alkyltrichloroacetimidates,[15] which can be isolated as stable and less hydrolysis-sensitive adducts.

With primary and secondary amines, N-substituted trichloroacetamidines are formed in a smooth reaction with good yields, which can be purified by vacuum distillation and are obtained as colorless, malodorous liquids.[16] Reaction with ammonia and then with anhydrous hydrogen chloride gives the solid trichloroacetamidine hydrochloride, the starting compound for the fungicide etridiazole.
In academic research, trichloroacetonitrile is used as a reagent in the Overman rearrangement, converting allylic alcohols into allylic amines.[17][18][19] The reaction is based on a [3,3]-sigmatropic and diastereoselective rearrangement.
Benzyl trichloroacetimidate is easily accessible from benzyl alcohol and trichloroacetonitrile.[20] Benzyl trichloroacetimidate is useful as a benzylating reagent for sensitive alcohols under mild conditions and to preserve chirality.[21]
O-Glycosyl-trichloroacetimidates for the activation of carbohydrates
R. R. Schmidt and co-workers[22] have described the selective anomeric activation of O-protected hexopyranoses (glucose, galactose, mannose, glucosamine, galactosamine), hexofuranoses and pentopyranoses with trichloroacetonitrile in the presence of a base, as well as glycosylations under acid catalysis.[23][24][25]
Under kinetic control[26] with potassium carbonate as the base, β-trichloroacetimidates are formed selectively, whereas with sodium hydride, cesium carbonate or potassium hydroxide[27] and in the presence of phase transfer catalysts[28] only α-trichloroacetimidates are obtained (thermodynamically controlled).
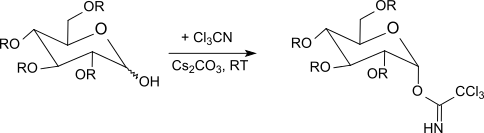
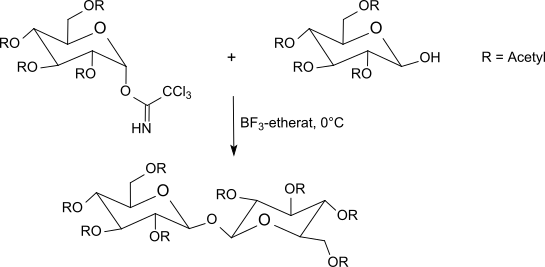
The trichloroacetimidates are reacted between -40 °C to room temperature with boron trifluoride etherate in dichloromethane with O-protected sugars. This method usually gives better results than the Koenigs-Knorr method using silver salts or the Helferich method which uses problematic mercury salts. Since an inversion occurs at the anomeric center, the reaction leads to β-O-glycosides (when using α-trichloroacetimidates). The trichloroacetimidate method often produces sterically uniform glycosides under mild reaction conditions in very good yields.
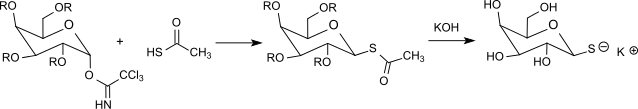
Thioacetic acid reacts with acetyl-protected α-galactosyl trichloroacetimidate even without additional acid catalysis to thioglycoside, from which (after cleavage of the protective groups) 1-thio-β-D-galactose is easily accessible, which is useful for the separation of racemates of amino acids.[29]
Trichloroacetonitrile was an important fumigant in the first half of the 20th century, but today it has become obsolete for this application.[30]
See also
References
- Peter Pollak, Gérard Romeder, Ferdinand Hagedorn, Heinz-Peter Gelbke "Nitriles" in Ullmann's Encyclopedia of Industrial Chemistry, 2002, Wiley-VCH, Weinheim. doi:10.1002/14356007.a17_363
- Bisschopinck, L. (1873). "Ueber die gechlorten Acetonitrile". Berichte der Deutschen Chemischen Gesellschaft. 6: 731–734. doi:10.1002/cber.187300601227.
- US 2375545, R. T. Foster, "Process for the preparation of trichloroacetonitrile", issued 1945-05-08, assigned to Imperial Chemical Industries
- US 2745868, G. Käbisch, "Process for the production of trichloroacetonitrile", issued 1956-05-15, assigned to Deutsche Gold- und Silber-Scheideanstalt, vormals Roessler
- Guidelines for Drinking Water Quality, 3. Auflage, Vol. 1, Recommendations, World Health Organization, Genf, 2004, ISBN 9-2415-4638-7, PDF.
- Frank Bernsdorff (2007), Untersuchungen zur abiotischen Bildung von Acetonitril, Haloacetonitrilen und Trichlornitromethan (in German), GRIN, p. 5, ISBN 9783638383431 }}
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Structure of Free Molecules in the Gas Phase, S. 9-46.
- E. D. Matveeva et al., Regioselective and stereoselective substitution of hydroxyl group for halogen in allyl alcohols, Zh. Org. Khim., 31, (8), 1121–1125 (1995).
- D. O. Jang et al., A mild and efficient procedure for the preparation of acid chlorides from carboxylic acids, Tetrahedron Lett., 40, (29), 5323–5326 (1999).
- J. Vago, J. Greiner, A useful acylation method using trichloroacetonitrile and triphenylphosphine for solid phase organic synthesis, Tetrahedron Lett., 43, (34), 6039–6041 (2002).
- O. Chantarasriwong et al., A practical and efficient method for the preparation of sulfonamides utilizing Cl3CCN/PPh3, Tetrahedron Lett., 47, (42), 7489–7492 (2006).
- A. Kasemsuknimit et al., Efficient amidation and esterification of phosphoric acid using Cl3CCN/Ph3P, Bull. Korean Chem. Soc., 32, (9), 3486–3488 (2011).
- W. Kijrungphaiboon et al., Cl3CCN/PPh3 and CBr4/PPh3: two efficient reagent systems for the preparation of N-heteroaromatic halides, Tetrahedron Lett., 53, 674–677 (2006).
- R. Martin (2011), Aromatic Hydroxyketones: Preparation and Physical Properties. Vol. 1 Hydroxybenzophenones (in German) (3. ed.), Springer, doi:10.1007/978-1-4020-9787-4, ISBN 978-1-4020-9787-4
- J. U. Nef, Ann. Chem., 287, 274 (1895).
- Grivas, John C.; Taurins, Alfred (1958-05-01). "Reaction of trichloroacetonitrile with primary and secondary amines: part i. preparation of some trichloroacetamidines". Canadian Journal of Chemistry. 36 (5): 771–774. doi:10.1139/v58-113. ISSN 0008-4042.
- T. Nishikawa; M. Asai; N. Ohyabu; M. Isobe (1998). "Improved Conditions for Facile Overman Rearrangement(1)". J. Org. Chem. 63 (1): 188–192. doi:10.1021/jo9713924. PMID 11674062.
- "Overman Rearrangement". Organic Chemistry Portal. organic-chemistry.org. Retrieved November 15, 2012.
- Y. K. Chen. A. E. Lurain, P. J. Walsh (2002). "A general, highly enantioselective method for the synthesis of D and L alpha-amino acids and allylic amines". J. Am. Chem. Soc. 124 (41): 12225–12231. doi:10.1021/ja027271p. PMID 12371863.
- Schaefer, Fred C.; Peters, Grace A. (1961). "Base-Catalyzed Reaction of Nitriles with Alcohols. A Convenient Route to Imidates and Amidine Salts". The Journal of Organic Chemistry. 26 (2): 412–418. doi:10.1021/jo01061a034.
- E. P. Eckenberg et al., A useful application of benzyl trichloroacetimidate for the benzylation of alcohols, Tetrahedron, 49, 1619–1624 (1993).
- R. R. Schmidt, J. Michel, Einfache Synthese von α- und β-O-Glycosylimidaten. Herstellung von Glykosiden und Disacchariden, Angew. Chem., 92, 763–764 (1980).
- R. R. Schmidt, Neue Methoden zur Glycosid- und Oligosaccharidsynthese – gibt es Alternativen zur Koenigs-Knorr-Methode?, Angew. Chem. 98, 213–236 (1986)
- R. R. Schmidt, W. Kinzy, Anomeric-oxygen activation for glycoside synthesis – the trichloroacetimidate method, Adv. Carbohydr. Chem. Biochem., 50, 21–123 (1994).
- R. R. Schmidt, K.-H. Jung, Oligosaccharide synthesis with trichloroacetimidates, In: Preparative Carbohydrate Chemistry, S. Hanessian, Ed., Marcel Dekker, New York, 283–312 (1997), ISBN 0-8247-9802-3.
- R. R. Schmidt, J. Michel, Liebigs Ann. Chem., 1343–1357 (1984).
- F. J. Urban et al., Tetrahedron Lett., 31, 4421–4424 (1990)
- V. J. Patil, Tetrahedron Lett., 37, 1481–1484 (1996).
- A. Jegorov et al., 1-Thio-β-D-galactose as a chiral derivatization agent for the resolution of D,L-aminoacid enantiomers, J. Chromatogr. A, 673(2), 286–290 (1994).
- N. M. Sax, R. J. Lewis, Hawley's Condensed Chemical Dictionary, 11th ed., Van Nostrand Reinhold, New York, S. 261, 1175 (1987).