Thermochemical cycle
Thermochemical cycles combine solely heat sources (thermo) with chemical reactions to split water into its hydrogen and oxygen components.[1] The term cycle is used because aside of water, hydrogen and oxygen, the chemical compounds used in these processes are continuously recycled.
If work is partially used as an input, the resulting thermochemical cycle is defined as a hybrid one.
History
This concept was first postulated by Funk and Reinstrom (1966) as a maximally efficient way to produce fuels (e.g. hydrogen, ammonia) from stable and abundant species (e.g. water, nitrogen) and heat sources.[2] Although fuel availability was scarcely considered before the oil crisis efficient fuel generation was an issue in important niche markets. As an example, in the military logistics field, providing fuels for vehicles in remote battlefields is a key task. Hence, a mobile production system based on a portable heat source (a nuclear reactor was considered) was being investigated with utmost interest. Following the oil crisis, multiple programs (Europe, Japan, USA) were created to design, test and qualify such processes for purposes such as energy independence. High temperature (1000K) nuclear reactors were still considered as the likely heat sources. However, optimistic expectations based on initial thermodynamics studies were quickly moderated by pragmatic analyses comparing standard technologies (thermodynamic cycles for electricity generation, coupled with the electrolysis of water) and by numerous practical issues (insufficient temperatures from even nuclear reactors, slow reactivities, reactor corrosion, significant losses of intermediate compounds with time...).[3] Hence, the interest for this technology faded during the next decades,[4] or at least some tradeoffs (hybrid versions) were being considered with the use of electricity as a fractional energy input instead of only heat for the reactions (e.g. Hybrid sulfur cycle). A rebirth in the year 2000 can be explained by both the new energy crisis, demand for electricity, and the rapid pace of development of concentrated solar power technologies whose potentially very high temperatures are ideal for thermochemical processes,[5] while the environmentally friendly side of thermochemical cycles attracted funding in a period concerned with a potential peak oil outcome.
Principles
Water-splitting via a single reaction
Consider a system composed of chemical species (e.g. water-splitting) in thermodynamic equilibrium at constant pressure and thermodynamic temperature T:
- H2O(l) H2(g) + 1/2 O2(g) (1)

Equilibrium is displaced to the right only if energy (enthalpy change ΔH for water-splitting) is provided to the system under strict conditions imposed by thermodynamics:
- one fraction must be provided as work, namely the Gibbs free energy change ΔG of the reaction: it consists of "noble" energy, i.e. under an organized state where matter can be controlled, such as electricity in the case of the electrolysis of water. Indeed, the generated electron flow can reduce protons (H+) ) at the cathode and oxidize anions (O2−) at the anode (the ions exist because of the chemical polarity of water), yielding the desired species.
- the other one must be supplied as heat, i.e. by increasing the thermal agitation of the species, and is equal by definition of the entropy to the absolute temperature T times the entropy change ΔS of the reaction.
- (2)

Hence, for an ambient temperature T° of 298K (kelvin) and a pressure of 1 atm (atmosphere (unit)) (ΔG° and ΔS° are respectively equal to 237 kJ/mol and 163 J/mol/K, relative to the initial amount of water), more than 80% of the required energy ΔH must be provided as work in order for water-splitting to proceed.
If phase transitions are neglected for simplicity's sake (e.g. water electrolysis under pressure to keep water in its liquid state), one can assume that ΔH et ΔS do not vary significantly for a given temperature change. These parameters are thus taken equal to their standard values ΔH° et ΔS° at temperature T°. Consequently, the work required at temperature T is,
- (3)

As ΔS° is positive, a temperature increase leads to a reduction of the required work. This is the basis of high-temperature electrolysis. This can also be intuitively explained graphically. Chemical species can have various excitation levels depending on the absolute temperature T, which is a measure of the thermal agitation. The latter causes shocks between atoms or molecules inside the closed system such that energy spreading among the excitation levels increases with time, and stop (equilibrium) only when most of the species have similar excitation levels (a molecule in a highly excited level will quickly return to a lower energy state by collisions) (Entropy (statistical thermodynamics)).
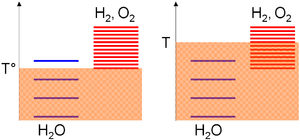
Relative to the absolute temperature scale, the excitation levels of the species are gathered based on standard enthalpy change of formation considerations; i.e. their stabilities. As this value is null for water but strictly positive for oxygen and hydrogen, most of the excitation levels of these last species are above the ones of water. Then, the density of the excitation levels for a given temperature range is monotonically increasing with the species entropy. A positive entropy change for water-splitting means far more excitation levels in the products. Consequently,
- A low temperature (T°), thermal agitation allow mostly the water molecules to be excited as hydrogen and oxygen levels required higher thermal agitation to be significantly populated (on the arbitrary diagram, 3 levels can be populated for water vs 1 for the oxygen/hydrogen subsystem),
- At high temperature (T), thermal agitation is sufficient for the oxygen/hydrogen subsystem excitation levels to be excited (on the arbitrary diagram, 4 levels can be populated for water vs 8 for the oxygen/hydrogen subsystem). According to the previous statements, the system will thus evolve toward the composition where most of its excitation levels are similar, i.e. a majority of oxygen and hydrogen species.
One can imagine that if T were high enough in Eq.(3), ΔG could be nullified, meaning that water-splitting would occur even without work (thermolysis of water). Though possible, this would require tremendously high temperatures: considering the same system naturally with steam instead of liquid water (ΔH° = 242 kJ/mol; ΔS° = 44 J/mol/K) would hence give required temperatures above 3000K, that make reactor design and operation extremely challenging.[6]
Hence, a single reaction only offers one freedom degree (T) to produce hydrogen and oxygen only from heat (though using Le Chatelier's principle would also allow to slightly decrease the thermolysis temperature, work must be provided in this case for extracting the gas products from the system)
Water-splitting with multiple reactions
On the contrary, as shown by Funk and Reinstrom, multiple reactions (e.g. k steps) provide additional means to allow spontaneous water-splitting without work thanks to different entropy changes ΔS°i for each reaction i. An extra benefit compared with water thermolysis is that oxygen and hydrogen are separately produced, avoiding complex separations at high temperatures.[7]
The first pre-requisites (Eqs.(4) and (5)) for multiple reactions i to be equivalent to water-splitting are trivial (cf. Hess's law):
- (4)

- (5)

Similarly, the work ΔG required by the process is the sum of each reaction work ΔGi:
- (6)

As Eq. (3) is a general law, it can be used anew to develop each ΔGi term. If the reactions with positive (p indice) and negative (n indice) entropy changes are expressed as separate summations, this gives,
- (7)

Using Eq. (6) for standard conditions allows to factorize the ΔG°i terms, yielding,
- (8)

Now consider the contribution of each summation in Eq. (8): in order to minimize ΔG, they must be as negative as possible:
- : -ΔS°i are negative, so (T-T°) must be as high as possible: hence, one choose to operate at the maximum process temperature TH
- : -ΔS°i are positive, (T-T°) should be ideally negative in order to decrease ΔG. Practically, one can only set T equals to T° as the minimum process temperature in order to get rid of this troublesome term (a process requiring a lower than standard temperature for energy production is a physical absurdity as it would require refrigerators and thus a higher work input than output). Consequently, Eq.(8) becomes,


- (9)

Finally, one can deduce from this last equation the relationship required for a null work requirement (ΔG ≤ 0)
- (10)

Consequently, a thermochemical cycle with i steps can be defined as sequence of i reactions equivalent to water-splitting and satisfying equations (4), (5) and (10). The key point to remember in that case is that the process temperature TH can theoretically be arbitrary chosen (1000K as a reference in most of the past studies, for high temperature nuclear reactors), far below the water thermolysis one.
This equation can alternatively (and naturally) be derived via the Carnot's theorem, that must be respected by the system composed of a thermochemical process coupled with a work producing unit (chemical species are thus in a closed loop):
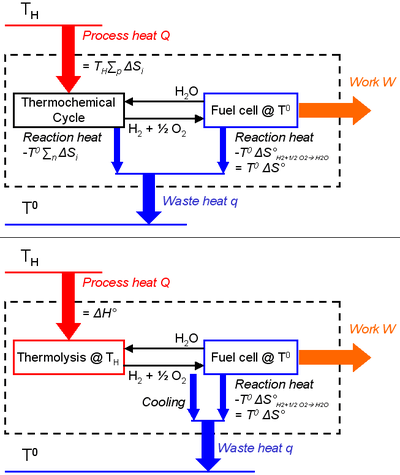
- at least two heat sources of different temperatures are required for cyclical operation, otherwise perpetual motion would be possible. This is trivial in the case of thermolysis, as the fuel is consumed via an inverse reaction. Consequently, if there is only one temperature (the thermolysis one), maximum work recovery in a fuel cell is equal to the opposite of the Gibbs free energy of the water-splitting reaction at the same temperature, i.e. null by definition of the thermolysis. Or differently said, a fuel is defined by its instability, so if the water/hydrogen/oxygen system only exists as hydrogen and oxygen (equilibrium state), combustion (engine) or use in a fuel cell would not be possible.
- endothermic reactions are chosen with positive entropy changes in order to be favored when the temperature increases, and the opposite for the exothermic reactions.
- maximal heat-to-work efficiency is the one of a Carnot heat engine with the same process conditions, i.e. a hot heat source at TH and a cold one at T°,
- (11)

- the work output W is the "noble" energy stored in the hydrogen and oxygen products (e.g. released as electricity during fuel consumption in a fuel cell). It thus corresponds to the free Gibbs energy change of water-splitting ΔG, and is maximum according to Eq.(3) at the lowest temperature of the process (T°) where it is equal to ΔG°.
- the heat input Q is the heat provided by the hot source at temperature TH to the i endothermic reactions of the thermochemical cycle (the fuel consumption subsystem is exothermic):
- (12)
- Hence, each heat requirement at temperature TH is,
- (13)
- Replacing Eq.(13) in Eq.(12) yields:
- (14)



Consequently, replacing W (ΔG°) and Q (Eq.(14)) in Eq.(11) gives after reorganization Eq.(10) (assuming that the ΔSi do not change significantly with the temperature, i.e. are equal to ΔS°i)
Equation (10) has practical implications about the minimum number of reactions for such a process according to the maximum process temperature TH.[8] Indeed, a numerical application (ΔG° equals to 229 kJ/K for water considered as steam) in the case of the originally chosen conditions (high-temperature nuclear reactor with TH and T° respectively equal to 1000K and 298K) gives a minimum value around 330 J/mol/K for the summation of the positive entropy changes ΔS°i of the process reactions.
This last value is very high as most of the reactions have entropy change values below 50 J/mol/K, and even an elevated one (e.g. water-splitting from liquid water: 163 J/mol/K) is twice lower. Consequently, thermochemical cycles composed of less than three steps are practically impossible with the originally planned heat sources (below 1000K), or require "hybrid" versions
Hybrid thermochemical cycles
In this case, an extra freedom degree is added via a relatively small work input Wadd (maximum work consumption, Eq.(9) with ΔG ≤ Wadd), and Eq.(10) becomes,
- (15)

If Wadd is expressed as a fraction f of the process heat Q (Eq.(14)), Eq.(15) becomes after reorganization,
- (16)

Using a work input equals to a fraction f of the heat input is equivalent relative to the choice of the reactions to operate a pure similar thermochemical cycle but with a hot source with a temperature increased by the same proportion f.
Naturally, this decreases the heat-to-work efficiency in the same proportion f. Consequently, if one want a process similar to a thermochemical cycle operating with a 2000K heat source (instead of 1000K), the maximum heat-to-work efficiency is twice lower. As real efficiencies are often significantly lower than ideal one, such a process is thus strongly limited.
Practically, use of work is restricted to key steps such as product separations, where techniques relying on work (e.g. electrolysis) might sometimes have fewer issues than those using only heat (e.g. distillations)
Particular case : Two-step thermochemical cycles
According to equation (10), the minimum required entropy change (right term) for the summation of the positive entropy changes decreases when TH increases. As an example, performing the same numerical application but with TH equals to 2000K would give a twice lower value (around 140 kJ/mol), which allows thermochemical cycles with only two reactions. Such processes can be realistically coupled with concentrated solar power technologies like Solar Updraft Tower. As an example in Europe, this is the goal of the Hydrosol-2 project (Greece, Germany (German Aerospace Center), Spain, Denmark, England) [9] and of the researches of the solar department of the ETH Zurich and the Paul Scherrer Institute (Switzerland).[10]
Examples of reactions satisfying high entropy changes are metal oxide dissociations, as the products have more excitation levels due to their gaseous state (metal vapors and oxygen) than the reactant (solid with crystalline structure, so symmetry dramatically reduces the number of different excitation levels). Consequently, these entropy changes can often be larger than the water-splitting one and thus a reaction with a negative entropy change is required in the thermochemical process so that Eq.(5) is satisfied. Furthermore, assuming similar stabilities of the reactant (ΔH°) for both thermolysis and oxide dissociation, a larger entropy change in the second case explained again a lower reaction temperature (Eq.(3)).
Let us assume two reactions, with positive (1 subscript, at TH) and negative (2 subscript, at T°) entropy changes. An extra property can be derived in order to have TH strictly lower than the thermolysis temperature: The standard thermodynamic values must be unevenly distributed among the reactions .[11]
Indeed, according to the general equations (2) (spontaneous reaction), (4) and (5), one must satisfy,
- (17)

Hence, if ΔH°1 is proportional to ΔH°2 by a given factor, and if ΔS°1 and ΔS°2 follow a similar law (same proportionality factor), the inequality (17) is broken (equality instead, so TH equals to the water thermolysis temperature).
Examples
Hundreds of such cycles have been proposed and investigated. This task has been eased by the availability of computers, allowing a systematic screening of chemical reactions sequences based on thermodynamic databases.[12] Only the main "families" will be described in this article.[13]
cycles with more than 3 steps or hybrid ones
Cycles based on the sulfur chemistry
An advantage of the sulfur chemical element is its high covalence. Indeed, it can form up to 6 chemical bonds with other elements such as oxygen (e.g. sulfates), i.e. a wide range of oxidation states. Hence, there exist several redox reactions involving such compounds. This freedom allows numerous chemical steps with different entropy changes, and thus offer more odds to meet the criteria required for a thermochemical cycle (cf. Principles). Most of the first studies were performed in the USA, as an example at the Kentucky University for sulfide-bases cycles.[14] Sulfate-based cycles were studied in the same laboratory [15] and also at Los Alamos National Laboratory [16] and at General Atomics. Significant researches based on sulfates (e.g. FeSO4 and CuSO4) were also performed in Germany [17] and in Japan .[18][19] However, the cycle which has given rise to the highest interests is probably the (Sulfur-iodine cycle) one (acronym: S-I) discovered by General Atomics.[20]
Cycles based on the reversed Deacon process
Above 973K, the Deacon reaction is reversed, yielding hydrogen chloride and oxygen from water and chlorine:
- H2O + Cl2 → 2 HCl + 1/2 O2
See also
- Iron oxide cycle
- Cerium(IV) oxide-cerium(III) oxide cycle
- Copper-chlorine cycle
- Hybrid sulfur cycle
- Hydrosol-2
- Sulfur-iodine cycle
- Zinc zinc-oxide cycle
- UT-3 cycle
References
- Producing Hydrogen: The Thermochemical Cycles - Idaho National Laboratory (INL)
- Funk, J.E., Reinstrom, R.M., 1966. Energy requirements in the production of hydrogen from water. I&EC Process Design and Development 5(3):336-342.
- Shinnar, R., Shapira, D., Zakai, S., 1981. Thermochemical and hybrid cycles for hydrogen production. A differential economic comparison with electrolysis. I&EC Process Design and Development 20(4):581-593.
- Funk, J.E., 2001. Thermochemical hydrogen production: past and present. International Journal of Hydrogen Energy 26(3):185:190.
- Steinfeld, A., 2005. Solar thermochemical production of hydrogen - a review. Solar Energy 78(5):603-615
- Lédé, J., Lapicque, F., Villermaux, J., Cales, B., Ounalli, A., Baumard, J.F., Anthony, A.M., 1982. Production of hydrogen by direct thermal decomposition of water: preliminary investigations. International Journal of Hydrogen Energy 7(12):939-950.
- Kogan, A., 1998. Direct solar thermal splitting of water and on-site separation of the products - II. Experimental feasibility study. International Journal of Hydrogen Energy 23(9):89-98.
- Abraham, B.M., Schreiner, F., 1974. General principles underlying chemical cycles which thermally decompose water into elements. I&EC Fundamentals 13(4):305-310.
- Roeb, M., Neises, M., Säck, J.P., Rietbrock, P., Monnerie, N.; Dersch, J., Schmitz, S., Sattler, C., 2009. Operational strategy of a two-step thermochemical process for solar hydrogen production. International Journal of Hydrogen Energy 34(10):4537-4545.
- Schunk, L.O., Lipinski, W., Steinfeld, A., 2009. Heat transfer model of a solar receiver-reactor for the thermal dissociation of ZnO – Experimental validation at 10 kW and scale-up to 1 MW. Chemical Engineering Journal 150(2-3):502-508.
- Glandt, E.D., Myers, A.L., 1976. Hydrogen production from water by means of chemical cycles. I&EC Process Design and Development 15(1):100-108.
- Russel, J.L., Porter, J.T., 1975. A search for thermochemical water-splitting cycles. Verziroglu, T.N., Hydrogen Energy, 517-529, Plenum
- Chao, R.E., 1974. Thermochemical water decomposition processes. I&EC Product Research Development 13(2):94-101.
- Ota K., Conger, W.L., 1977. Thermochemical hydrogen production via a cycle using barium and sulfur : reaction between barium sulfide and water. International Journal of Hydrogen Energy 2(2):101:106.
- Soliman, M.A., Conger, W.L., Carty, R.H., Funk, J.E., Cox, K.E., 1976. Hydrogen production via thermochemical cycles based on sulfur chemistry. International Journal of Hydrogen Energy 1(3):265-270.
- Mason, C.F.m 1977. The reduction of hydrogen bromide using transition metal compounds. International Journal of hydrogen energy 1(4):427-434.
- Schulten, R.m Knoche, K.F., Erzeugung von Wasserstoff und Sauerstoff aus Wasser mit Hilfe von Wärme. German Patent #2 257 103, December, the 26th, 1974
- Yoshida, K., Kameyama, H., Toguchi, K., 1975. Proceedings of the U.S. Japan Joint Seminar Publication Office Ohta's Laboratory Yokohama National University, Tokyo, 20–23 June
- Kameyama, H., Yoshida, K., Kunii, D., 1976. A method for screening possible thermochemical decomposition processes for water using deltaG-T diagrams. The Chemical Engineering Journal 11(3):223-229.
- Besenbruch, G. 1982. General Atomic sulfur iodine thermochemical water-splitting process. Proceedings of the American Chemical Society, Div. Pet. Chem., 27(1):48-53.