Stereoselectivity
In chemistry, stereoselectivity[1] is the property of a chemical reaction in which a single reactant forms an unequal mixture of stereoisomers during a non-stereospecific creation of a new stereocenter or during a non-stereospecific transformation of a pre-existing one.[2] The selectivity arises from differences in steric effects and electronic effects in the mechanistic pathways leading to the different products. Stereoselectivity can vary in degree but it can never be total since the activation energy difference between the two pathways is finite. Both products are at least possible and merely differ in amount. However, in favorable cases, the minor stereoisomer may not be detectable by the analytic methods used.
An enantioselective reaction is one in which one enantiomer is formed in preference to the other, in a reaction that creates an optically active product from an achiral starting material, using either a chiral catalyst, an enzyme or a chiral reagent. The degree of selectivity is measured by the enantiomeric excess. An important variant is kinetic resolution, in which a pre-existing chiral center undergoes reaction with a chiral catalyst, an enzyme or a chiral reagent such that one enantiomer reacts faster than the other and leaves behind the less reactive enantiomer, or in which a pre-existing chiral center influences the reactivity of a reaction center elsewhere in the same molecule.
A diastereoselective reaction is one in which one diastereomer is formed in preference to another (or in which a subset of all possible diastereomers dominates the product mixture), establishing a preferred relative stereochemistry. In this case, either two or more chiral centers are formed at once such that one relative stereochemistry is favored,[3] or a pre-existing chiral center (which needs not be optically pure) biases the stereochemical outcome during the creation of another. The degree of relative selectivity is measured by the diastereomeric excess.
Stereoconvergence can be considered an opposite of stereospecificity, when the reaction of two different stereoisomers yield a single product stereoisomer.
The quality of stereoselectivity is concerned solely with the products, and their stereochemistry. Of a number of possible stereoisomeric products, the reaction selects one or two to be formed.
Examples
An example of modest stereoselectivity is the dehydrohalogenation of 2-iodo-butane which yields 60% trans-2-butene and 20% cis-2-butene.[4] Since alkene geometric isomers are also classified as diastereomers, this reaction would also be called diastereoselective.
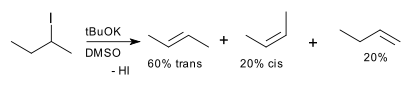
The addition of formic acid to norbornene is also stereospecific because the exo isomer is formed exclusively without any of the endo isomer:[5]
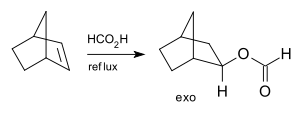
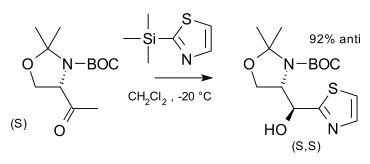
Cram's rule predicts the major diastereomer resulting from the diastereoselective nucleophilic addition to a carbonyl group next to a chiral center. The chiral center need not be optically pure, as the relative stereochemistry will be the same for both enantiomers. In the example below the (S)-aldehyde reacts with a thiazole to form the (S,S) diastereomer but only a small amount of the (S,R) diastereomer:[6]
The Sharpless epoxidation is an example of an enantioselective process, in which an achiral allylic alcohol substrate is transformed into an optically active epoxyalcohol. In the case of chiral allylic alcohols, kinetic resolution results. Another example is Sharpless asymmetric dihydroxylation. In the example below the achiral alkene yields only one of possible 4 stereoisomers.[7]
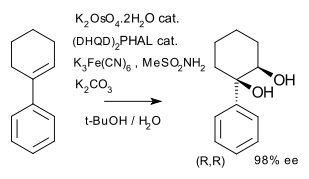
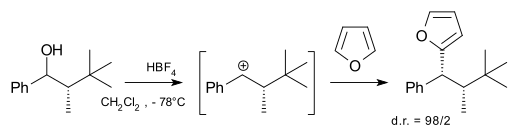
With a stereogenic center next to the carbocation the substitution can be stereoselective in inter- [8] and intramolecular [9][10] reactions. In the reaction depicted below the nucleophile (furan) can approach the carbocation formed from the least shielded side away from the bulky t-butyl group resulting in high facial diastereoselectivity:
Stereoselective biosynthesis
Pinoresinol biosynthesis involved a protein called a dirigent protein. The first dirigent protein was discovered in Forsythia intermedia. This protein has been found to direct the stereoselective biosynthesis of (+)-pinoresinol from coniferyl alcohol monomers.[11] Recently, a second, enantiocomplementary dirigent protein was identified in Arabidopsis thaliana, which directs enantioselective synthesis of (−)-pinoresinol.[12]
-Pinoresinol_Biosynthesis.svg.png)
Notes and references
- (a)"Overlap Control of Carbanionoid Reactions. I. Stereoselectivity in Alkaline Epoxidation," Zimmerman, H. E.; Singer, L.; Thyagarajan, B. S. J. Am. Chem. Soc., 1959, 81, 108-116. (b)Eliel, E., "Stereochemistry of Carbon Compound", McGraw-Hill, 1962 pp 434-436.
- For instance, the SN1 reaction destroys a pre-existing stereocenter then creates a new one.
- Or fewer than all possible relative stereochemistries are obtained.
- Effects of base strength and size upon in base-promoted elimination reactions. Richard A. Bartsch, Gerald M. Pruss, Bruce A. Bushaw, Karl E. Wiegers J. Am. Chem. Soc.; 1973; 95(10); 3405-3407. doi:10.1021/ja00791a067
- Organic Syntheses Coll. Vol. 5, p.852 (1973); Vol. 42, p.79 (1962). Link
- Organic Syntheses, Coll. Vol. 10, p.140 (2004); Vol. 77, p.78 (2000). Link
- Organic Syntheses, Coll. Vol. 10, p.603 (2004); Vol. 79, p.93 (2002).Link
- Diastereoselective Friedel-Crafts Cyclization Reactions to 2-Substituted 1-Phenyl-1,2,3,4-tetrahydronaphthalenes Friedrich Mühlthau, Thorsten Bach Synthesis 2005: 3428-3436 doi:10.1055/s-2005-918482
- High Facial Diastereoselectivity in Intra- and Intermolecular Reactions of Chiral Benzylic Cations Friedrich Mühlthau, Oliver Schuster, and Thorsten Bach J. Am. Chem. Soc., 2005, 127 (26), pp 9348–9349 doi:10.1021/ja050626v
- Stereoselective Reactions with Stabilized Carbocations Pier Giorgio Cozzi and Fides Benfatti Angew. Chem. Int. Ed. 2009, 48 doi:10.1002/anie.200905235
- Davin LB, Wang HB, Crowell AL, et al. (1997). "Stereoselective bimolecular phenoxy radical coupling by an auxiliary (dirigent) protein without an active center". Science. 275 (5298): 362–6. doi:10.1126/science.275.5298.362. PMID 8994027.
- Pickel B, Constantin M-A, Pfannsteil J, Conrad J, Beifuss U, Schaffer A (March 2007). "An Enantiocomplementary Dirigent Protein for the Enantioselective Laccase-Catalyzed Oxidative Coupling of Phenols". Angewandte Chemie. 53 (4): 273–284. doi:10.1007/s10086-007-0892-x.