Pharmacophore
A pharmacophore is an abstract description of molecular features that are necessary for molecular recognition of a ligand by a biological macromolecule. IUPAC defines a pharmacophore to be "an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response".[1] A pharmacophore model explains how structurally diverse ligands can bind to a common receptor site. Furthermore, pharmacophore models can be used to identify through de novo design or virtual screening novel ligands that will bind to the same receptor.
Features
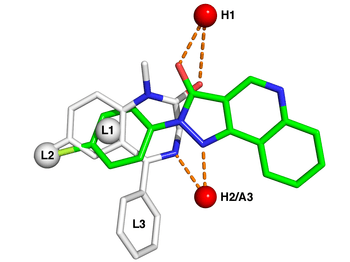
Typical pharmacophore features include hydrophobic centroids, aromatic rings, hydrogen bond acceptors or donors, cations, and anions. These pharmacophoric points may be located on the ligand itself or may be projected points presumed to be located in the receptor.
The features need to match different chemical groups with similar properties, in order to identify novel ligands. Ligand-receptor interactions are typically “polar positive”, “polar negative” or “hydrophobic”. A well-defined pharmacophore model includes both hydrophobic volumes and hydrogen bond vectors.
Model development
The process for developing a pharmacophore model generally involves the following steps:
- Select a training set of ligands – Choose a structurally diverse set of molecules that will be used for developing the pharmacophore model. As a pharmacophore model should be able to discriminate between molecules with and without bioactivity, the set of molecules should include both active and inactive compounds.
- Conformational analysis – Generate a set of low energy conformations that is likely to contain the bioactive conformation for each of the selected molecules.
- Molecular superimposition – Superimpose ("fit") all combinations of the low-energy conformations of the molecules. Similar (bioisosteric) functional groups common to all molecules in the set might be fitted (e.g., phenyl rings or carboxylic acid groups). The set of conformations (one conformation from each active molecule) that results in the best fit is presumed to be the active conformation.
- Abstraction – Transform the superimposed molecules into an abstract representation. For example, superimposed phenyl rings might be referred to more conceptually as an 'aromatic ring' pharmacophore element. Likewise, hydroxy groups could be designated as a 'hydrogen-bond donor/acceptor' pharmacophore element.
- Validation – A pharmacophore model is a hypothesis accounting for the observed biological activities of a set of molecules that bind to a common biological target. The model is only valid insofar as it is able to account for differences in biological activity of a range of molecules.
As the biological activities of new molecules become available, the pharmacophore model can be updated to further refine it.
Applications
In modern computational chemistry, pharmacophores are used to define the essential features of one or more molecules with the same biological activity. A database of diverse chemical compounds can then be searched for more molecules which share the same features arranged in the same relative orientation. Pharmacophores are also used as the starting point for developing 3D-QSAR models. Such tools and a related concept of "privileged structures", which are "defined as molecular frameworks which are able of providing useful ligands for more than one type of receptor or enzyme target by judicious structural modifications",[3] aid in drug discovery.[4]
History
Historically, pharmacophores were established by Lemont Kier, who first mentions the concept in 1967[5] and uses the term in a publication in 1971.[6]
The development of the concept is often erroneously accredited to Paul Ehrlich. However neither the alleged source[7] nor any of his other works mention the term "pharmacophore" or make use of the concept.[8]
See also
- Cheminformatics
- Molecule mining
- Pharmaceutical company
- QSAR
- in silico
References
- Wermuth CG, Ganellin CR, Lindberg P, Mitscher LA (1998). "Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998)". Pure and Applied Chemistry. 70 (5): 1129–1143. doi:10.1351/pac199870051129.
- Madsen U, Bräuner-Osborne H, Greenwood JR, Johansen TN, Krogsgaard-Larsen P, Liljefors T, Nielsen M, Frølund B (2005). "GABA and Glutamate receptor ligands and their therapeutic potential in CNS disorders". In Gad SC (ed.). Drug Discovery Handbook. Hoboken, N.J: Wiley-Interscience/J. Wiley. pp. 797–907. ISBN 0-471-21384-5.
- Duarte, CD; et al. (2007), "Privileged structures: a useful concept for the rational design of new lead drug candidates", Mini Rev Med Chem, 7 (11): 1108–1119, doi:10.2174/138955707782331722, PMID 18045214.
- Jangampalli Adi, Pradeepkiran (February 2019). "Pharmacophore-based models for therapeutic drugs against phosphorylated tau in Alzheimer's disease". Drug Discovery Today. 24 (2): 616–623. doi:10.1016/j.drudis.2018.11.005. PMC 6397090. PMID 30453058.
- Kier LB (September 1967). "Molecular orbital calculation of preferred conformations of acetylcholine, muscarine, and muscarone". Mol. Pharmacol. 3 (5): 487–94. PMID 6052710.
- Kier LB (1971). Molecular orbital theory in drug research. Boston: Academic Press. pp. 164–169. ISBN 0-12-406550-3.
- Ehrlich P (1909). "Über den jetzigen Stand der Chemotherapie". Ber. Dtsch. Chem. Ges. 42: 17–47. doi:10.1002/cber.19090420105.
- J.H. van Drie (2007). "Monty Kier and the Origin of the Pharmacophore Concept" (PDF). Internet Electronic Journal of Molecular Design. 6: 271–279.CS1 maint: uses authors parameter (link)
Further reading
- Güner OF, ed. (1999). Pharmacophore perception, development, and use in drug design. LaJolla, CA: International University Line. ISBN 0-9636817-6-1.
- Langer T, Hoffmann RD (2006). Pharmacophores and pharmacophore searches. Weinheim: WILEY-VCH. ISBN 3-527-31250-1.
External links
The following computer software packages enable the user to model the pharmacophore using a variety of computational chemistry methods: