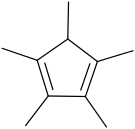
Pentamethylcyclopentadiene
1,2,3,4,5-Pentamethylcyclopentadiene is a cyclic dialkene with the formula C5Me5H (Me = CH3).[1] 1,2,3,4,5-Pentamethylcyclopentadiene is the precursor to the ligand 1,2,3,4,5-pentamethylcyclopentadienyl, which is often denoted Cp* (C5Me5) and read as "C P star", the "star" signifying the five methyl groups radiating from the core of the ligand. In contrast to less-substituted cyclopentadiene derivatives, Cp*H is not prone to dimerization.
 | |
 | |
| Identifiers | |
|---|---|
3D model (JSmol) |
|
| ChemSpider | |
| ECHA InfoCard | 100.021.586 |
PubChem CID |
|
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| C10H16 | |
| Molar mass | 136.24 g/mol |
| Boiling point | 55 to 60 °C (131 to 140 °F; 328 to 333 K) at 13 mmHg (1.7 kPa) |
| Sparingly soluble | |
| Hazards | |
| Flash point | 114 °C (237 °F; 387 K) |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
Synthesis
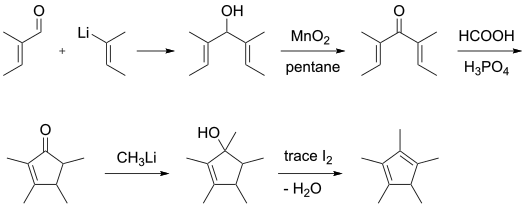
Pentamethylcyclopentadiene is commercially available. It was first prepared from tiglaldehyde via 2,3,4,5-tetramethylcyclopent-2-enone.[2]
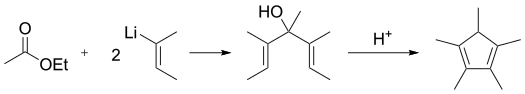
Alternatively, 2-butenyllithium adds to ethyl acetate followed by acid-catalyzed dehydrocyclization:[3][4]

Organometallic derivatives
Cp*H is a precursor to organometallic compounds containing the C
5Me−
5 ligand, commonly called Cp*−.[5]
| Cp*–metal complexes | |
|---|---|
| Cp*2Fe | yellow |
| Cp*TiCl3 | red |
| [Cp*Fe(CO)2]2 | red-violet |
| [Cp*RhCl2]2 | red |
| Cp*IrCl22 | orange |
| Cp*Re(CO)3 | colorless |
| Cp*Mo(CO)2CH3 | orange |
Some representative reactions leading to such Cp*–metal complexes follow:[6]
Some Cp* complexes are prepared using silyl transfer:
- Cp*Li + Me3SiCl → Cp*SiMe3 + LiCl
- Cp*SiMe3 + TiCl4 → Cp*TiCl3 + Me3SiCl
Some Cp* complexes were originally prepared using hexamethyl Dewar benzene as the precursor. This method was traditionally used for [Rh(C5Me5)Cl2]2, but has been discontinued with the increased commercial availability of Cp*H.
- 2 Cp*H + 2 Fe(CO)5 → [η5-Cp*Fe(CO)2]2 + H2 + 6 CO
For the related Cp complex, see cyclopentadienyliron dicarbonyl dimer.
An instructive but obsolete route to Cp* complexes involves the use of hexamethyl Dewar benzene. This method was traditionally used for preparation of the chloro-bridged dimers [Cp*IrCl2]2 and [Cp*RhCl2]2. Such syntheses rely on a hydrohalic acid induced rearrangement of hexamethyl Dewar benzene[7][8] to a substituted pentamethylcyclopentadiene prior to reaction with the hydrate of either iridium(III) chloride[9] or rhodium(III) chloride.[10]

Comparison to other Cp ligands
Complexes of pentamethylcyclopentadienyl differ in several ways from the more common cyclopentadienyl (Cp) derivatives. Being more electron-rich, Cp*− is a stronger donor and dissociation, like ring-slippage, is more difficult with Cp* than with Cp.[11] The fluorinated ligand, (trifluoromethyl)tetramethylcyclopentadienyl, C5Me4CF3, combines the properties of Cp and Cp*: it possesses the steric bulk of Cp* but has electronic properties similar to Cp, the electron-donation from the methyl groups being "canceled out" by the electron-accepting nature of the trifluoromethyl substituent.[12] Its steric bulk stabilizes complexes with fragile ligands. Its bulk also attenuates intermolecular interactions, decreasing the tendency to form polymeric structures. Its complexes also tend to be more soluble in non-polar solvents. The methyl group in Cp* complexes can undergo C–H activation leading to "tuck-in complexes". Bulky cyclopentadienyl ligands are known that are far more sterically encumbered than Cp*.
3C5H3.png)
See also
References
- Elschenbroich, C.; Salzer, A. (1989). Organometallics: A Concise Introduction. VCH. p. 47. ISBN 9783527278183.
- De Vries, L. (1960). "Preparation of 1,2,3,4,5-Pentamethyl-cyclopentadiene, 1,2,3,4,5,5-Hexamethyl-cyclopentadiene, and 1,2,3,4,5-Pentamethyl-cyclopentadienylcarbinol". J. Org. Chem. 25 (10): 1838. doi:10.1021/jo01080a623.
- Threlkel, S.; Bercaw, J. E.; Seidler, P. F.; Stryker, J. M.; Bergman, R. G. (1993). "1,2,3,4,5-Pentamethylcyclopentadiene". Organic Syntheses.; Collective Volume, 8, p. 505
- Fendrick, C. M.; Schertz, L. D.; Mintz, E. A.; Marks, T. J. (1992). Large-Scale Synthesis of 1,2,3,4,5-Pentamethylcyclopentadiene. Inorganic Syntheses. 29. pp. 193–198. doi:10.1002/9780470132609.ch47. ISBN 978-0-470-13260-9.
- Yamamoto, A. (1986). Organotransition Metal Chemistry: Fundamental Concepts and Applications. Wiley-Interscience. p. 105. ISBN 9780471891710.
- King, R. B.; Bisnette, M. B. (1967). "Organometallic chemistry of the transition metals XXI. Some π-pentamethylcyclopentadienyl derivatives of various transition metals". J. Organomet. Chem. 8 (2): 287–297. doi:10.1016/S0022-328X(00)91042-8.
- Paquette, L. A.; Krow, G. R. (1968). "Electrophilic Additions to Hexamethyldewarbenzene". Tetrahedron Lett. 9 (17): 2139–2142. doi:10.1016/S0040-4039(00)89761-0.
- Criegee, R.; Gruner, H. (1968). "Acid-catalyzed Rearrangements of Hexamethyl-prismane and Hexamethyl-Dewar-benzene". Angew. Chem. Int. Ed. Engl. 7 (6): 467–468. doi:10.1002/anie.196804672.
- Kang, J. W.; Mosley, K.; Maitlis, P. M. (1968). "Mechanisms of Reactions of Dewar Hexamethylbenzene with Rhodium and Iridium Chlorides". Chem. Commun. (21): 1304–1305. doi:10.1039/C19680001304.
- Kang, J. W.; Maitlis, P. M. (1968). "Conversion of Dewar Hexamethylbenzene to Pentamethylcyclopentadienylrhodium(III) Chloride". J. Am. Chem. Soc. 90 (12): 3259–3261. doi:10.1021/ja01014a063.
- Kuwabara, Takuya; Tezuka, Ryogen; Ishikawa, Mikiya; Yamazaki, Takuya; Kodama, Shintaro; Ishii, Youichi (2018-06-25). "Ring Slippage and Dissociation of Pentamethylcyclopentadienyl Ligand in an (η 5 -Cp*)Ir Complex with a κ 3 - O , C , O Tridentate Calix[4]arene Ligand under Mild Conditions". Organometallics. 37 (12): 1829–1832. doi:10.1021/acs.organomet.8b00257. ISSN 0276-7333.
- Gassman, Paul G.; Mickelson, John W.; Sowa, John R. (1992-08-01). "1,2,3,4-Tetramethyl-5-(trifluoromethyl)cyclopentadienide: a unique ligand with the steric properties of pentamethylcyclopentadienide and the electronic properties of cyclopentadienide". Journal of the American Chemical Society. 114 (17): 6942–6944. doi:10.1021/ja00043a065. ISSN 0002-7863.