Management of thalassemia
Treatment of the inherited blood disorder thalassemia depends upon the level of severity. For mild forms of the condition, advice and counseling are often all that are necessary. For more severe forms, treatment may consist in blood transfusion; chelation therapy to reverse iron overload, using drugs such as deferoxamine, deferiprone, or deferasirox; medication with the antioxidant indicaxanthin to prevent the breakdown of hemoglobin; or a bone marrow transplant using material from a compatible donor, or from the patient's mother. Removal of the spleen (splenectomy) could theoretically help to reduce the need for blood transfusions in people with thalassaemia major or intermedia but there is currently no reliable evidence from clinical trials about its effects.[1] Population screening has had some success as a preventive measure.
Levels of severity
- Mild thalassemia : patients with thalassemia traits do not require medical or follow-up care after the initial diagnosis is made.[2] Patients with β-thalassemia trait should be warned that their condition can be misdiagnosed for the common Iron deficiency anemia. They should eschew empirical use of Iron therapy; yet iron deficiency can develop during pregnancy or from chronic bleeding.[3] Counseling is indicated in all persons with genetic disorders, especially when the family is at risk of a severe form of disease that may be prevented.[4]
- Severe thalassemia : Patients with severe thalassemia require medical treatment. A blood transfusion regimen was the first measure effective in prolonging life.[2]
Medication
Patients with thalassemia gradually accumulate high levels of iron (Fe) in their bodies. This build-up of iron may be due to the disease itself, from irregular hemoglobin not properly incorporating adequate iron into its structure, or it may be due to the many blood transfusions received by the patient. This overload of iron brings with it many biochemical complications.
Two key substances involved in iron transport and storage in the body are ferritin and transferrin. Ferritin is a protein present within cells that binds to Fe (II) and stores it as Fe (III), releasing it into the blood whenever required. Transferrin is an iron-binding protein present in blood plasma; transferrin acts as a transporter, carrying iron through blood and providing cells with the metal through endocytosis. Transferrin is highly specific to iron (III), and binds to it with an equilibrium constant of 1023 M−1 at a pH of 7.4.[5]
Thalassemia results in nontransferrin-bound iron being available in blood as all the transferrin becomes fully saturated. This free iron is toxic to the body since it catalyzes reactions that generate free hydroxyl radicals.[6] These radicals may induce lipid peroxidation of organelles like lysosomes, mitochondria, and sarcoplasmic membranes. The resulting lipid peroxides may interact with other molecules to form cross links, and thus either cause these compounds to perform their functions poorly, or render them non-functional altogether.[6] This iron overload may be treated with chelation therapy. Deferoxamine, deferiprone and deferasirox are the three most widely used iron-chelating agents.
Deferoxamine
Structure and coordination
The drug deferoxamine, also known as desferoxamine B and DFO-B, is a trihydroxamic acid that is produced by the actinobacteria Streptomyces pilosus. It binds iron, decreasing the toxic reactions catalysed by the unbound metal, and it also decreases the uptake of iron by tissues. Deferoxamine achieves this by acting as a hexadentate iron-chelating ligand: it binds to all six coordination sites on nontransferrin-bound iron, effectively deactivating it.[7] Deferoxamine is mostly specific to ferric iron (Fe3+) and coordinates to Fe3+ using the oxygen atoms on its multiplehydroxyl and carbonyl groups, forming a structure called ferrioxamine. This drug-iron complex is mostly excreted by the kidneys as it is water-soluble.[8] Approximately one-third of ferrioxamine could also be excreted through the feces in bile.[6]
Administration and action
Deferoxamine is administered via intravenous, intramuscular, or subcutaneous injections. Oral administration is not possible as deferoxamine is rapidly metabolized by enzymes and is poorly absorbed from the gastrointestinal tract. The required parenteral administration represents one of deferoxamine’s downfalls as it is harder for patients to follow up with their therapy due to the financial and emotional burdens experienced.[9] Deferoxamine was proven to cure many clinical complications and diseases that result from iron overload. It beneficially affects cardiac disease, such as myocardial disease which occurs as a result of iron accumulation in the heart.[10] Deferoxamine was also shown to improve liver function by arresting the development of hepatic fibrosis which occurs as a result of iron accumulation in the liver.[11] Deferoxamine also has positive effects on endocrine function and growth. Endocrine abnormalities in thalassemic patients involve the overloaded iron interfering with the production of insulin-like growth factor (IGF-1), as well as stimulating hypogonadism, both of which cause poor pubertal growth. A study showed that 90% of patients who were regularly treated with deferoxamine since childhood had normal pubertal growth, which fell to 38% for patients treated only with low doses of deferoxamine since their teens.[6] Another endocrine abnormality that thalassemic patients face is diabetes mellitus, which results from iron overload in the pancreas impairing insulin secretion. Studies have shown that patients who were regularly treated with deferoxamine have a reduced risk of developing diabetes mellitus.[12]
Side effects
Deferoxamine could lead to toxic side effects if doses greater than 50 mg/kg body weight are administered. These side effects may include auditory and ocular abnormalities, pulmonary toxicity, sensorimotor neurotoxicity, as well as changes in renal function.[6] Another toxic effect of deferoxamine mostly observed in children is the failure of linear growth. This reduction in height may occur as a result of deferoxamine chelating metals other than iron which are required for normal growth. Deferoxamine has an affinity constant (Ka) of 1031 for Fe3+, 1014 for Cu2+ and 1010 for Zn2+, and so may coordinate to zinc and copper when little iron is available for chelation. Zinc is needed for the proper functioning of various metalloenzymes involved in bone formation. Zinc chelation may cause zinc deficiency in the body, which can thus lead to a reduced growth rate, reduced collagen formation and defective bone mineralization. Similarly, copper functions as an enzyme cofactor in bone formation. Copper chelation may result in copper deficiency as well, leading to metaphyseal cupping and osteoporosis. For example, abnormal collagen is formed when copper is deficient as the enzyme lysyl oxidase, which uses copper as a cofactor and catalyzes the oxidative deamination step that is important for cross-linking of collagen, cannot function properly. Studies have shown that even though the blood serum of patients receiving deferoxamine was not deficient in copper and zinc, deficiencies of the metals in the metaphyseal matrix were observed.
The toxic effect of deferoxamine on linear growth could also be due to excess deferoxamine accumulating in tissues and interfering with iron-dependent enzymes which are involved in the post-translational modification of collagen.[13]
Patients who receive vitamin C supplements have shown improved iron excretion by deferoxamine. This occurs due to the expansion of the iron pool brought about by vitamin C, which deferoxamine subsequently has access to. However, vitamin C supplementation could also worsen iron toxicity by promoting the formation of free radicals. Therefore, only 100 mg of vitamin C should be taken 30 minutes to one hour after deferoxamine administration.[14]
It has also been proven that combined treatment with deferoxamine and deferiprone leads to an increased efficiency in chelation and doubles iron excretion.[15]
Deferiprone
Structure and coordination
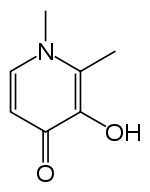
Deferiprone (DFP) is a bidentate iron-chelator. Three molecules of the drug therefore coordinate to one iron atom, forming an orthorhombic structure.[16]
DFP is synthetically made and is highly selective to Fe(III).[16][17] Physical properties that allow this compound to be effective as a drug include its water solubility, low molecular weight (139 Da), neutral charge, and lipophilicity.[16] These physio-chemical properties allow facile crossing of cell membranes throughout the body, including the blood-brain barrier, facilitating removal of excess iron from within organs.[16][18]
Although the mechanism for the removal of iron by DFP is not well understood, however, a study by Viroj Wiwanitkit in 2006 proposed a possible mechanism: the coordination to the iron was thought to occur through the cleavage of either a C-C bond or a C-O bond in the drug. Wiwanitkit concluded that the mechanism goes through the cleavage of the C-C bond because this bond requires less energy to be cleaved. The total energy for the cleavage was found to be negative, suggesting spontaneity and thermodynamic favourability of the cleavage. The resulting structure of the product also resembled the observed tertiary structureof the drug-iron complex.[19]
Administration and action
Deferiprone is an iron chelator that is orally active, its administration thus being much easier than that for deferoxamine.[16] Plasma levels for the iron-drug complex climax after one hour of intake and the drug has a half-life of 160 minutes. Most of the iron-drug complex is therefore excreted within three to four hours following administration, the excretion occurring mostly in urine (90%).[16]
When comparing deferiprone to deferoxamine, they both bind iron with similar efficiency. However, drugs with different properties are able to access different iron pools. DFP is smaller than deferoxamine and can thus enter cells more easily. Also, at the pH of blood, the affinity of DFP for iron is concentration dependent: at low DFP concentrations, the iron-drug complex breaks down and the iron is donated to another competing ligand. This property accounts for the observed tendency of DFP to redistribute iron in the body. For the same reason, DFP can ‘shuttle’ intracellular iron out to the plasma, and transfer the iron to deferoxamine which goes on to expel it from the body.[17]
DFP was also found to be significantly more effective than deferoxamine in treating myocardial siderosis in patients with thalassemia major:[16] DFP is thought to improve the function of mitochondria in the heart by accessing and redistributing labile iron in cardiac cells.
Thalassemia patients may also be faced with potential oxidative damage to brain cells as the brain has high oxygen demands, but contains relatively low levels of antioxidant agents for protection against oxidation. The presence of excess iron in the brain may lead to higher concentrations of free radicals. Hexadentate chelators, like deferoxamine, are large molecules, and are thus unlikely to be able to cross the blood-brain barrier to chelate the excess iron. DFP, however, can do so and forms a soluble, neutral iron-drug complex that can cross cell membranes by non-facilitated diffusion. Attaching the drug to sugars may additionally enhance the penetration of the blood-brain barrier, as the brain uses facilitated transport for its relatively high levels of sugar intake.[20]
Side effects
DFP can be subjected to glucuronidation in the liver, which may expel as much as 85% of the drug from the body before it has had a chance to chelate iron. DFP also has a well-known safety profile, with agranulocytosis being the most serious side effect.[16] While agranulocytosis has been reported in less than 2% of patients treated, it is potentially life-threatening and thus requires close monitoring of the white blood cell count.[18] Less serious side effects include gastrointestinal symptoms, which were found in 33% of patients in the first year of administration, but fell to 3% in following years; arthralgia; and zinc deficiency, with the latter being a problem especially for individuals with diabetes.[16]
Deferasirox
Structure and coordination
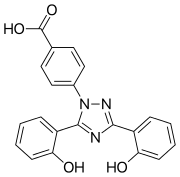
Deferasirox is an N-substituted bis-hydroxyphenyl-triazole. It is capable of removing iron from the blood through the coordination of two molecules of the deferasirox to a single iron ion, which forms the iron chelate (Fe-[deferasirox]2).[21] Each molecule of the tridentate chelator deferasirox binds to the iron at three sites, using one nitrogen atom and two oxygen atoms. This results in a stable octahedral geometry around the iron centre. The ability of deferasirox to remove iron stems directly from its relatively small size, which is what allows it to access the iron contained within the blood and inside tissues. Also, an important feature of deferasirox is that it has been shown to be highly selective for iron in the +3 oxidation state, and use of the drug does not lead to a significant decrease in the levels of other important metals in the body.[22]
Administration and action
_complex.png)
Deferasirox is most commonly marketed under the brand name Exjade. It has one key advantage over desferoxamine in that it can be taken orally in pill form, and so does not require intravenous or subcutaneous administration. With a terminal elimination half life of 8–16 hours, the deferasirox pill can be taken just once everyday. A once-daily dose of 20 mg/kg of body weight has been found to be sufficient for most patients for the maintenance of liver iron concentration (LIC) levels, which are usually measured as mg of iron per g of liver tissue. Larger doses may be required for some patients in order to reduce LIC levels.[23] The ability of deferasirox to effectively reduce LIC levels has been well documented. One study demonstrated that after 4–5 years of deferasirox treatment the mean LIC levels of patients decreased from 17.4 ± 10.5 to 9.6 ± 8.0 mg Fe/g. This study showed that long-term treatment did result in a sustainable reduction in the iron burden faced by patients receiving blood transfusions for thalassemia.[24] An additional benefit of the use of deferasirox instead of desferoxamine is that, unlike desferoxamine, early studies have indicated that deferasirox does not have a significant impact on the growth and development of pediatric thalassemia patients. In a study by Cappellini et al. it was shown that children receiving the treatment displayed continual near-normal growth and development over a 5-year study period.[24]
Side effects
Deferasirox can, however, have a wide variety of side effects. These may include headaches, nausea, vomiting, and joint pains.[25] Some evidence has been shown of a link to gastrointestinal disorders experienced by some people who have received the treatment.[24]
Indicaxanthin
Structure
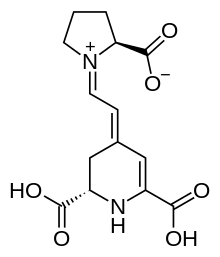
Indicaxanthin is a pigment derived from the cactus pear fruit and can be used as an antioxidant. Dietary indicaxanthin has been shown to have protective effects on RBCs in people with beta thalassemia.[26] It has a structure similar to that of amino acids, and is amphiphilic: it is able to bind to cell membranes through charge-related interactions with polar head groups of membrane constituents, as well through adsorption to the lipid aggregates. Upon ex vivo introduction to thalassemic blood, indicaxanthin was shown to accumulate within RBCs.[26]
Function
Hb undergoes the following oxidation reaction during normal controlled breakdown of RBCs:
Hb → Oxy-Hb → Met-Hb → [Perferryl-Hb] → Oxoferryl → further oxidation steps
This reaction is experienced by thalassemic RBCs to a greater extent because, not only are there more oxidative radicals in thalassemic blood, but thalassemic RBCs also have limited antioxidant defense. Indicaxanthin is able to reduce the perferryl-Hb, a reactive intermediate, back to met-Hb. The overall effect of this step is that Hb degradation is prevented, which helps prevent accelerated breakdown of RBCs.[26]
In addition, indicaxathin has been shown to reduce oxidative damage in cells and tissues and does so by binding to radicals. The mechanism of its function, however, is still unknown.[26]
Indicaxanthin has high bioavailability and minimal side effects, like vomiting or diarrhea.
Carrier detection
- A screening policy exists in Cyprus to reduce the incidence of thalassemia, which since the program's implementation in the 1970s (which also includes pre-natal screening and abortion) has reduced the number of children born with the hereditary blood disease from 1 out of every 158 births to almost zero.[27]
- In Iran as a premarital screening, the man's red cell indices are checked first, if he has microcytosis (mean cell hemoglobin < 27 pg or mean red cell volume < 80 fl), the woman is tested. When both are microcytic their hemoglobin A2 concentrations are measured. If both have a concentration above 3.5% (diagnostic of thalassemia trait) they are referred to the local designated health post for genetic counseling.[28]
In 2008, in Spain, a baby was selectively implanted in order to be a cure for his brother's thalassemia. The child was born from an embryo screened to be free of the disease before implantation with In vitro fertilization. The baby's supply of immunologically compatible cord blood was saved for transplantation to his brother. The transplantation was considered successful.[29] In 2009, a group of doctors and specialists in Chennai and Coimbatore registered the successful treatment of thalassemia in a child using a sibling's umbilical cord blood.[30]
Bone marrow transplant
It is possible to be cured, by a bone marrow transplantation (BMT) from compatible donor. In low-risk young people, the thalassemia-free survival rate is 87%; the mortality risk is 3%.[31] The drawback is that this method requires an HLA-matched compatible donor.
If the person does not have an HLA-matched compatible donor, there is another method called bone marrow transplantation from haploidentical mother to child (mismatched donor), in which the donor is the mother. The results are these: thalassemia-free survival rate 70%, rejection 23%, and mortality 7%. The best results are with very young people.[32]
References
- Sharma, A; Easow Mathew, M; Puri, L (17 September 2019). "Splenectomy for people with thalassaemia major or intermedia". The Cochrane Database of Systematic Reviews. 9: CD010517. doi:10.1002/14651858.CD010517.pub3. PMC 6746994. PMID 31529486.
- "Pediatric Thalassemia Treatment & Management". Medical Care. Open Publishing. 30 April 2010. Retrieved 27 September 2011.
- Claude Owen Burdick. "Separating Thalassemia Trait and Iron Deficiency by Simple Inspection". American Society for Clinical Pathology. Archived from the original on 22 September 2014. Retrieved 27 September 2011.
- Harrison's Principles of Internal Medicine 17th Edition. McGraw-Hill medical. September 2008. p. 776. ISBN 978-0-07-164114-2.
- Aisen P, Leibman A, Zweier J; Leibman; Zweier (March 1978). "Stoichiometric and site characteristics of the binding of iron to human transferrin". J. Biol. Chem. 253 (6): 1930–7. PMID 204636.CS1 maint: multiple names: authors list (link)
- >Brittenham, Gary M; Olivieri, Nancy F (1997). "Iron-chelating therapy and the treatment of thalassemia". Journal of the American Society of Hematology. 89 (3): 739–761. PMID 9028304. Retrieved 28 February 2013.
- Brittenham, Gary M.; Griffith, Patricia M.; Nienhuis, Arthur W.; McLaren, Christine E.; Young, Neal S.; Tucker, Eben E.; Allen, Christopher J.; Farrell, David E.; Harris, John W. (1994). "Efficacy of Deferoxamine in Preventing Complications of Iron Overload in Patients with Thalassemia Major". New England Journal of Medicine. 331 (9): 567–73. doi:10.1056/NEJM199409013310902. PMID 8047080.
- Cozar, O.; Leopold, N.; Jelic, C.; Chiş, V.; David, L.; Mocanu, A.; Tomoaia-Cotişel, M. (2006). "IR, Raman and surface-enhanced Raman study of desferrioxamine B and its Fe(III) complex, ferrioxamine B". Journal of Molecular Structure. 788 (1–3): 1–6. Bibcode:2006JMoSt.788....1C. doi:10.1016/j.molstruc.2005.04.035.
- Cohen, Alan; Martin, Marie; Schwartz, Elias (1981). "Response to long-term deferoxamine therapy in thalassemia". The Journal of Pediatrics. 99 (5): 689–94. doi:10.1016/S0022-3476(81)80385-X. PMID 7299539.
- Pennell, D. J.; Berdoukas, V; Karagiorga, M; Ladis, V; Piga, A; Aessopos, A; Gotsis, ED; Tanner, MA; et al. (2006). "Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis". Blood. 107 (9): 3738–44. doi:10.1182/blood-2005-07-2948. PMID 16352815.
- Jin, Haiyan; Terai, Shuji; Sakaida, Isao (2007). "The iron chelator deferoxamine causes activated hepatic stellate cells to become quiescent and to undergo apoptosis". Journal of Gastroenterology. 42 (6): 475–84. doi:10.1007/s00535-007-2020-5. PMID 17671763.
- Kaye, Todd B.; Guay, André T.; Simonson, Donald C. (1993). "Non-insulin-dependent diabetes mellitus and elevated serum ferritin level". Journal of Diabetes and Its Complications. 7 (4): 245–249. doi:10.1016/S0002-9610(05)80252-1.
- Olivieri, Nancy F.; Koren, Gideon; Harris, Jonathan; Khattak, Sohail; Freedman, Melvin H.; Templeton, Douglas M.; Bailey, John D.; Reilly, B. J. (1992). "Growth Failure and Bony Changes Induced by Deferoxamine". Journal of Pediatric Hematology/Oncology. 14: 48–56. doi:10.1097/00043426-199221000-00007. PMID 1550263.
- Ambruso, DR; Mahony, BS; Githens, JH; Rhoades, ED (1982). "Effect of subcutaneous deferoxamine and oral vitamin C on iron excretion in congenital hypoplastic anemia and refractory anemia associated with the 5q-syndrome". The American Journal of Pediatric Hematology/Oncology. 4 (2): 115–23. PMID 7114394.
- Kattamis, Antonis (2005). "Combined therapy with deferoxamine and deferiprone". Annals of the New York Academy of Sciences. 1054 (1): 175–82. Bibcode:2005NYASA1054..175K. doi:10.1196/annals.1345.020. PMID 16339663.
- Wiwanitkit, Viroj (2006). "Quantum chemical analysis of the deferiprone-iron binding reaction". International Journal of Nanomedicine. 1 (1): 111–3. doi:10.2147/nano.2006.1.1.111. PMC 2426763. PMID 17722270.
- Olivieri, Nancy F.; Brittenham, Gary M. (1997). "Iron-Chelating Therapy and the Treatment of Thalassemia". Blood. 89 (3): 739–61. doi:10.1182/blood.V89.3.739. PMID 9028304.
- Galanello, R.; Campus, S. (2009). "Deferiprone Chelation Therapy for Thalassemia Major". Acta Haematologica. 122 (2–3): 155–64. doi:10.1159/000243800. PMID 19907153.
- Wiwanitkit, Viroj (2006). "Quantum Chemical Analysis of the Deferiprone-Iron Binding Reaction". International Journal of Nanomedicine. 1 (1): 111–3. doi:10.2147/nano.2006.1.1.111. PMC 2426763. PMID 17722270.
- Heli, Hossein; Mirtorabi, Siamak; Karimian, Khashayar (2011). "Advances in iron chelation: An update". Expert Opinion on Therapeutic Patents. 21 (6): 819–56. doi:10.1517/13543776.2011.569493. PMID 21449664.
- Cappellini, M. D.; Cohen, A; Piga, A; Bejaoui, M; Perrotta, S; Agaoglu, L; Aydinok, Y; Kattamis, A; et al. (2006). "A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia". Blood. 107 (9): 3455–62. doi:10.1182/blood-2005-08-3430. PMID 16352812.
- R Galanello; Piga, A; Forni, GL; Bertrand, Y; Foschini, ML; Bordone, E; Leoni, G; Lavagetto, A; et al. (2006-01-01). "Phase II clinical evaluation of deferasirox, a once-daily oral chelating agent, in pediatric patients with β-thalassemia major". Haematologica. 91 (10): 1343–51. PMID 17018383.
- Nisbet-Brown, Eric; Olivieri, Nancy F; Giardina, Patricia J; Grady, Robert W; Neufeld, Ellis J; Séchaud, Romain; Krebs-Brown, Axel J; Anderson, Judith R; et al. (2003). "Effectiveness and safety of ICL670 in iron-loaded patients with thalassaemia: A randomised, double-blind, placebo-controlled, dose-escalation trial". The Lancet. 361 (9369): 1597–602. doi:10.1016/S0140-6736(03)13309-0. PMID 12747879.
- Cappellini, M. D.; Bejaoui, M.; Agaoglu, L.; Canatan, D.; Capra, M.; Cohen, A.; Drelichman, G.; Economou, M.; et al. (2011). "Iron chelation with deferasirox in adult and pediatric patients with thalassemia major: Efficacy and safety during 5 years' follow-up". Blood. 118 (4): 884–93. doi:10.1182/blood-2010-11-316646. PMID 21628399.
- "How Are Thalassemias Treated?". National Heat, Lung and Blood institute. Retrieved March 2, 2013.
- Tesoriere, L.; Allegra, M.; Butera, D.; Gentile, C.; Livrea, M. A. (2006). "Cytoprotective effects of the antioxidant phytochemical indicaxanthin in β-thalassemia red blood cells". Free Radical Research. 40 (7): 753–61. doi:10.1080/10715760600554228. PMID 16984002.
- Leung TN, Lau TK, Chung TKh; Lau; Chung (April 2005). "Thalassaemia screening in pregnancy". Current Opinion in Obstetrics and Gynecology. 17 (2): 129–34. doi:10.1097/01.gco.0000162180.22984.a3. PMID 15758603.CS1 maint: multiple names: authors list (link)
- Samavat A, Modell B; Modell (November 2004). "Iranian national thalassaemia screening programme". BMJ (Clinical Research Ed.). 329 (7475): 1134–7. doi:10.1136/bmj.329.7475.1134. PMC 527686. PMID 15539666.
- Spanish Baby Engineered To Cure Brother
- His sister's keeper: Brother's blood is boon of life, Times of India, 17 September 2009
- Sabloff, M; Chandy, M; Wang, Z; Logan, BR; Ghavamzadeh, A; Li, CK; Irfan, SM; Bredeson, CN; et al. (2011). "HLA-matched sibling bone marrow transplantation for β-thalassemia major". Blood. 117 (5): 1745–50. doi:10.1182/blood-2010-09-306829. PMC 3056598. PMID 21119108.
- Sodani, P; Isgrò, A; Gaziev, J; Paciaroni, K; Marziali, M; Simone, MD; Roveda, A; De Angelis, G; et al. (2011). "T cell-depleted hla-haploidentical stem cell transplantation in thalassemia young patients". Pediatric Reports. 3 Suppl 2 (Suppl 2): e13. doi:10.4081/pr.2011.s2.e13. PMC 3206538. PMID 22053275.