Magnetic circular dichroism
Magnetic circular dichroism (MCD) is the differential absorption of left and right circularly polarized (LCP and RCP) light, induced in a sample by a strong magnetic field oriented parallel to the direction of light propagation. MCD measurements can detect transitions which are too weak to be seen in conventional optical absorption spectra, and it can be used to distinguish between overlapping transitions. Paramagnetic systems are common analytes, as their near-degenerate magnetic sublevels provide strong MCD intensity that varies with both field strength and sample temperature. The MCD signal also provides insight into the symmetry of the electronic levels of the studied systems, such as metal ion sites.[1]
History
It was first shown by Faraday that optical activity (the Faraday effect) could be induced in matter by a longitudinal magnetic field (a field in the direction of light propagation).[2] The development of MCD really began in the 1930s when a quantum mechanical theory of MOR (magnetic optical rotatory dispersion) in regions outside absorption bands was formulated. The expansion of the theory to include MCD and MOR effects in the region of absorptions, which were referred to as “anomalous dispersions” was developed soon thereafter. There was, however, little effort made to refine MCD as a modern spectroscopic technique until the early 1960s. Since that time there have been numerous studies of MCD spectra for a very large variety of samples, including stable molecules in solutions, in isotropic solids, and in the gas phase, as well as unstable molecules entrapped in noble gas matrices. More recently, MCD has found useful application in the study of biologically important systems including metalloenzymes and proteins containing metal centers.[3][4]
Differences between CD and MCD
In natural optical activity, the difference between the LCP light and the RCP light is caused by the asymmetry of the molecules. Because of the handedness of the molecule, the absorption of the LCP light would be different from the RCP light. However, in MCD in the presence of a magnetic field, LCP and RCP no longer interact equivalently with the absorbing medium. Thus there is not the same direct relation between magnetic optical activity and molecular stereochemistry which would be expected, because it is found in natural optical activity. So, natural CD is much more rare than MCD.[5]
Although there is much overlap in the requirements and use of instruments, ordinary CD instruments are usually optimized for operation in the ultraviolet, approximately 170–300 nm, while MCD instruments are typically required to operate in the visible to near infrared, approximately 300–2000 nm. The physical processes that lead to MCD are substantively different from those of CD. However, like CD, it is dependent on the differential absorption of left and right hand circularly polarized light. MCD will only exist at a given wavelength if the studied sample has an optical absorption at that wavelength.[1] This is distinctly different from the related phenomenon of optical rotatory dispersion (ORD), which can be observed at wavelengths far from any absorption band.
Measurement
The MCD signal ΔA is derived via the absorption of the LCP and RCP light as

This signal is often presented as a function of wavelength λ, temperature T or magnetic field H.[1] MCD spectrometers can simultaneously measure absorbance and ΔA along the same light path.[6] This eliminates error introduced through multiple measurements or different instruments that previously occurred before this advent. The MCD spectrometer example shown below begins with a light source that emits a monochromatic wave of light. This wave is passed through a Rochon prism linear polarizer, which separates the incident wave into two beams that are linearly polarized by 90 degrees. The two beams follow different paths- one beam (the extraordinary beam) traveling directly to a photomultiplier (PMT), and the other beam (the ordinary beam) passing through a photoelastic modulator (PEM) oriented at 45 degrees to the direction of the ordinary ray polarization. The PMT for the extraordinary beam detects the light intensity of the input beam. The PEM is adjusted to cause an alternating plus and minus 1/4 wavelength shift of one of the two orthogonal components of the ordinary beam. This modulation converts the linearly polarized light into circularly polarized light at the peaks of the modulation cycle. Linearly polarized light can be decomposed into two circular components with intensity represented as

The PEM will delay one component of linearly polarized light with a time dependence that advances the other component by 1/4 λ (hence, quarter-wave shift). The departing circularly polarized light oscillates between RCP and LCP in a sinusoidal time-dependence as depicted below:
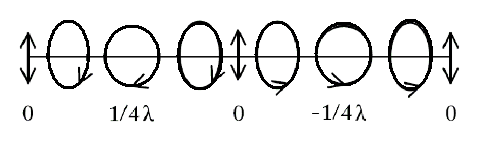
The light finally travels through a magnet containing the sample, and the transmittance is recorded by another PMT. The schematic is given below:
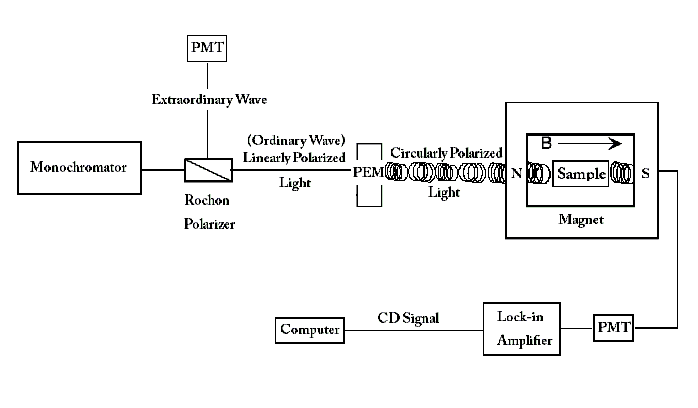
The intensity of light from the ordinary wave that reaches the PMT is governed by the equation:

Here A– and A+ are the absorbances of LCP or RCP, respectively; ω is the modulator frequency – usually a high acoustic frequency such as 50 kHz; t is time; and δ0 is the time-dependent wavelength shift.
This intensity of light passing through the sample is converted into a two-component voltage via a current/voltage amplifier. A DC voltage will emerge corresponding to the intensity of light passed through the sample. If there is a ΔA, then a small AC voltage will be present that corresponds to the modulation frequency, ω. This voltage is detected by the lock in amplifier, which receives its reference frequency, ω, directly from the PEM. From such voltage, ΔA and A can be derived using the following relations:


where Vex is the (DC) voltage measured by the PMT from the extraordinary wave, and Vdc is the DC component of the voltage measured by the PMT for the ordinary wave (measurement path not shown in the diagram).
Some superconducting magnets have a small sample chamber, far too small to contain the entire optical system. Instead, the magnet sample chamber has windows on two opposite sides. Light from the source enters one side, interacts with the sample (usually also temperature controlled) in the magnetic field, and exits through the opposite window to the detector. Optical relay systems that allow the source and detector each to be about a meter from the sample are typically employed. This arrangement avoids many of the difficulties that would be encountered if the optical apparatus had to operate in the high magnetic field, and also allows for a much less expensive magnet.
Applications
MCD can be used as an optical technique for the detection of electronic structure of both the ground states and excited states. It is also a strong addition to the more commonly used absorption spectroscopy, and there are two reasons that explain this. First, a transition buried under a stronger transition can appear in MCD if the first derivative of the absorption is much larger for the weaker transition or it is of the opposite sign. Second, MCD will be found where no absorption is detected at all if ΔA > (ΔAmin) but A<Amin, where (ΔA)min and Amin are the minimum of ΔA and A that are detectable. Typically, (ΔAmin) and Amin are of the magnitudes around 10−5 and 10−3 respectively. So, a transition can only be detected in MCD, not in the absorption spectroscopy, if ΔA/A > 10−2. This happens in paramagnetic systems that are at lower temperature or that have sharp lines in the spectroscopy.[7]
In biology, metalloproteins are the most likely candidates for MCD measurements, as the presence of metals with degenerate energy levels leads to strong MCD signals. In the case of ferric heme proteins,[8] MCD is capable of determining both oxidation and spin state to a remarkably exquisite degree. In regular proteins, MCD is capable of stoichiometrically measuring the tryptophan content of proteins, assuming there are no other competing absorbers in the spectroscopic system. In addition, the application of MCD spectroscopy greatly improved the level of understanding in the ferrous non-heme systems because of the direct observation of the d–d transitions, which generally can not be obtained in optical absorption spectroscopy owing to the weak extinction coefficients and are often electron paramagnetic resonance silent due to relatively large ground-state sublevel splittings and fast relaxation times.[9]
Theory
Consider a system of localized, non-interacting absorbing centers. Based on the semi-classical radiation absorption theory within the electric dipole approximation, the electric vector of the circularly polarized waves propagates along the +z direction. In this system, is the angular frequency, and = n – ik is the complex refractive index. As the light travels, the attenuation of the beam is expressed as[7]



where is the intensity of light at position , is the absorption coefficient of the medium in the direction, and is the speed of light. Circular dichroism (CD) is then defined by the difference between left () and right () circularly polarized light, , following the sign convention of natural optical activity. In the presence of a static, uniform external magnetic field applied parallel to the direction of propagation of light,[2] the Hamiltonian for the absorbing center takes the form for describing the system in the external magnetic field and describing the applied electromagnetic radiation. The absorption coefficient for a transition between two eigenstates of , and , can be described using the electric dipole transition operator as















The term is a frequency-independent correction factor allowing for the effect of the medium on the light wave electric field, composed of the permittivity and the real refractive index .



Discrete line spectrum
In cases of a discrete spectrum, the observed at a particular frequency can be treated as a sum of contributions from each transition,



where is the contribution at from the transition, is the absorption coefficient for the transition, and is a bandshape function (). Because eigenstates and depend on the applied external field, the value of varies with field. It is frequently useful to compare this value to the absorption coefficient in the absence of an applied field, often denoted







When the Zeeman effect is small compared to zero-field state separations, line width, and and when the line shape is independent of the applied external field , first-order perturbation theory can be applied to separate into three contributing Faraday terms, called , , and . The subscript indicates the moment such that contributes a derivative-shaped signal and and contribute regular absorptions. Additionally, a zero-field absorption term is defined. The relationships between , , and these Faraday terms are









for external field strength , Boltzmann constant , temperature , and a proportionality constant . This expression requires assumptions that is sufficiently high in energy that , and that the temperature of the sample is high enough that magnetic saturation does not produce nonlinear term behavior. Though one must pay attention to proportionality constants, there is a proportionality between and molar extinction coefficient and absorbance for concentration and path length .









These Faraday terms are the usual language in which MCD spectra are discussed. Their definitions from perturbation theory are[10]

where is the degeneracy of ground state , labels states other than or , and and label the levels within states and and (respectively), is the energy of unperturbed state , is the angular momentum operator, is the spin operator, and indicates the real part of the expression.











Origins of A, B, and C Faraday Terms

The equations in the previous subsection reveal that the , , and terms originate through three distinct mechanisms.
The term arises from Zeeman splitting of the ground or excited degenerate states. These field-dependent changes in energies of the magnetic sublevels causes small shifts in the bands to higher/lower energy. The slight offsets result in incomplete cancellation of the positive and negative features, giving a net derivative shape in the spectrum. This intensity mechanism is generally independent of sample temperature.
The term is due to the field-induced mixing of states. Energetic proximity of a third state to either the ground state or excited state gives appreciable Zeeman coupling in the presence of an applied external field. As the strength of the magnetic field increases, the amount of mixing increases to give growth of an absorption band shape. Like the term, the term is generally temperature independent. Temperature dependence of term intensity can sometimes be observed when is particularly low-lying in energy.



The term requires the degeneracy of the ground state, often encountered for paramagnetic samples. This happens due to a change in the Boltzmann population of the magnetic sublevels, which is dependent on the degree of field-induced splitting of the sublevel energies and on the sample temperature.[11] Decrease of the temperature and increase of the magnetic field increases the term intensity until it reaches the maximum (saturation limit). Experimentally, the term spectrum can be obtained from MCD raw data by subtraction of MCD spectra measured in the same applied magnetic field at different temperatures, while and terms can be distinguished via their different band shapes.[9]
The relative contributions of A, B and C terms to the MCD spectrum are proportional to the inverse line width, energy splitting, and temperature:

where is line width and is the zero-field state separation. For typical values of = 1000 cm−1, = 10,000 cm−1 and = 6 cm−1 (at 10 K), the three terms make relative contributions 1:0.1:150. So, at low temperature the term dominates over and for paramagnetic samples.[12]


Example on C terms
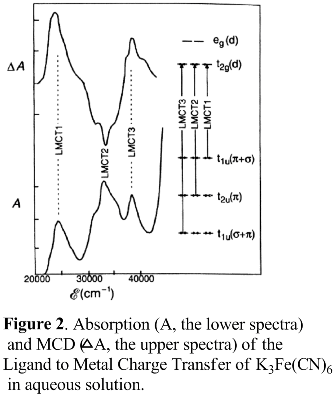
In the visible and near-ultraviolet regions, the hexacyanoferrate(III) ion (Fe(CN)63−) exhibits three strong absorptions at 24500, 32700, and 40500 cm−1, which have been ascribed to ligand to metal charge transfer (LMCT) transitions. They all have lower energy than the lowest-energy intense band for the Fe(II) complex Fe(CN)62− found at 46000 cm−1.[13] The red shift with increasing oxidation state of the metal is characteristic of LMCT bands.
These features can be explained as follows. The ground state of the anion is 2T2g, which derives from the electronic configuration (t2g)5. So, there would be an unpaired electron in the d orbital of Fe3+ From that, the three bands can be assigned to the transitions 2t2g→2t1u1, 2t2g →2t1u2, 2t2g →2t2u. Two of the excited states are of the same symmetry, and, based on the group theory, they could mix with each other so that there are no pure σ and π characters in the two t1u states, but for t2u, there would be no intermixing. The A terms are also possible from the degenerate excited states, but the studies of temperature dependence showed that the A terms are not as dependent as the C term.[14]
An MCD study of Fe(CN)63− embedded in a thin polyvinyl alcohol (PVA) film revealed a temperature dependence of the C term. The room-temperature C0/D0 values for the three bands in the Fe(CN)63− spectrum are 1.2, −0.6, and 0.6, respectively, and their signs (positive, negative, and positive) establish the energy ordering as 2t2g→2t1u2<2t2g→2t2u<2t2g→2t1u1
Example on A and B terms
To have an A- and B-term in the MCD spectrum, a molecule must contain degenerate excited states (A-term) and excited states close enough in energy to allow mixing (B-term). One case exemplifying these conditions is a square planar, d8 complex such as [(n-C4H9)4N]2Pt(CN)4. In addition to containing A- and B-terms, this example demonstrates the effects of spin-orbit coupling in metal to ligand charge transfer (MLCT) transitions. As shown in figure 1, the molecular orbital diagram of [(n-C4H9)4N]2Pt(CN)4 reveals MLCT into the antibonding π* orbitals of cyanide. The ground state is diamagnetic (thereby eliminating any C-terms) and the LUMO is the a2u. The dipole-allowed MLCT transitions are a1g-a2u and eg-a2u. Another transition, b2u-a2u, is a weak (orbitally forbidden singlet) but can still be observed in MCD.[15]

Because A- and B-terms arise from the properties of states, all singlet and triplet excited states are given in figure 2.


Mixing of all these singlet and triplet states will occur and is attributed to the spin orbit coupling of platinum 5d orbitals (ζ ~ 3500 cm−1), as shown in figure 3. The black lines on the figure indicate the mixing of 1A2u with 3Eu to give two A2u states. The red lines show the 1Eu, 3Eu, 3A2u, and 3B1u states mixing to give four Eu states. The blue lines indicate remnant orbitals after spin-orbit coupling that are not a result of mixing.
References
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "magnetic circular dichroism". doi:10.1351/goldbook.MT06778
- A. D. Buckingham & P. J. Stephens (1966). "Magnetic Optical Activity". Annu. Rev. Phys. Chem. 17: 399. Bibcode:1966ARPC...17..399B. doi:10.1146/annurev.pc.17.100166.002151.
- W. Roy Mason (2007). A practical guide to magnetic circular dichroism spectroscopy. Wiley-Interscience. doi:10.1002/9780470139233. ISBN 978-0-470-06978-3. Retrieved 16 April 2011.
- P. N. Schatz; A. J. McCafferyd (1969). "The Faraday effect". Quarterly Reviews, Chemical Society. 23 (4): 552. doi:10.1039/QR9692300552.
- Dennis Caldwell; Thorne, J M; Eyring, H (1971). "Magnetic Circular Dichroism". Annu. Rev. Phys. Chem. 22: 259–278. Bibcode:1971ARPC...22..259C. doi:10.1146/annurev.pc.22.100171.001355.
- G. A. Osborne (1973). "A Near-Infrared Circular Dichroism and Magnetic Circular Dichroism Instrument". Review of Scientific Instruments. 44: 10. Bibcode:1973RScI...44...10O. doi:10.1063/1.1685944.
- Stephens, P. J. (1974). "Magnetic Circular Dichroism". Annu. Rev. Phys. Chem. 25: 201. Bibcode:1974ARPC...25..201S. doi:10.1146/annurev.pc.25.100174.001221.
- G. Zoppellaro; et al. (2009). "Review: Studies of ferric heme proteins with highly anisotropic/highly axial low spin (S = 1/2) electron paramagnetic resonance signals with bis-Histidine and histidine-methionine axial iron coordination". Biopolymers. 91 (12): 1064–82. doi:10.1002/bip.21267. PMC 2852197. PMID 19536822.
- E.I. Solomon; et al. (1995). "Magnetic circular dichroism spectroscopy as a probe of the geometric and electronic structure of non-heme ferrous enzymes". Coordination Chemistry Reviews. 144: 369–460. doi:10.1016/0010-8545(95)01150-N.
- Stephens, P. J. (1976). "Magnetic Circular Dichroism". Adv. Chem. Phys. 35: 197. doi:10.1002/9780470142547.ch4.
- Lehnert, N.; DeBeer George, S.; Solomon, E. I. (2001). "Recent advances in bioinorganic spectroscopy". Current Opinion in Chemical Biology. 5 (2): 176–187. doi:10.1016/S1367-5931(00)00188-5. PMID 11282345.
- Neese, F.; Solomon, E. I. (1999). "MCD C-Term Signs, Saturation Behavior, and Determination of Band Polarizations in Randomly Oriented Systems with Spin S >/= (1)/(2). Applications to S = (1)/(2) and S = (5)/(2)". Inorg. Chem. 38 (8): 1847–1865. doi:10.1021/ic981264d. PMID 11670957.
- Stephens, P. J. (1965). "The Faraday Rotation of Allowed Transitions: Charge-Transfer Transitions in K3Fe(CN)6". Inorg. Chem. 4 (12): 1690–1692. doi:10.1021/ic50034a003.
- Upton, A. H. P.; Williamson, B. E. (1994). "Magnetic circular dichroism and absorption spectra of hexacyanoferrate(III) in a poly(vinyl alcohol) film". J. Phys. Chem. 98: 71. doi:10.1021/j100052a013.
- Isci, H.; Mason, W. R. (1975). "Electronic structure and spectra of square-planar cyano and cyanoamine complexes of platinum(II)". Inorg. Chem. 14 (4): 905. doi:10.1021/ic50146a038.