Lysosome
A lysosome (/ˈlaɪsəˌsoʊm/) is a membrane-bound organelle found in many animal cells.[1] They are spherical vesicles that contain hydrolytic enzymes that can break down many kinds of biomolecules. A lysosome has a specific composition, of both its membrane proteins, and its lumenal proteins. The lumen's pH (~4.5–5.0)[2] is optimal for the enzymes involved in hydrolysis, analogous to the activity of the stomach. Besides degradation of polymers, the lysosome is involved in various cell processes, including secretion, plasma membrane repair, apoptosis, cell signaling, and energy metabolism.[3]
| Cell biology | |
|---|---|
| The animal cell | |
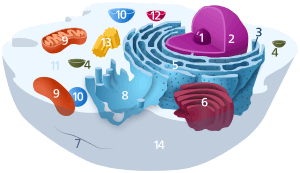 Components of a typical animal cell:
|
Lysosomes act as the waste disposal system of the cell by digesting in use materials in the cytoplasm, from both inside and outside the cell. Material from outside the cell is taken-up through endocytosis, while material from the inside of the cell is digested through autophagy.[5] The sizes of the organelles vary greatly—the larger ones can be more than 10 times the size of the smaller ones.[6] They were discovered and named by Belgian biologist Christian de Duve, who eventually received the Nobel Prize in Physiology or Medicine in 1974.
Lysosomes are known to contain more than 60 different enzymes, and have more than 50 membrane proteins.[7][8] Enzymes of the lysosomes are synthesised in the rough endoplasmic reticulum and exported to the Golgi apparatus upon recruitment by a complex composed of CLN6 and CLN8 proteins.[9][10] The enzymes are trafficked from the Golgi apparatus to lysosomes in small vesicles, which fuse with larger acidic vesicles. Enzymes destined for a lysosome are specifically tagged with the molecule mannose 6-phosphate, so that they are properly sorted into acidified vesicles.[11][12]
In 2009, Marco Sardiello and coworkers discovered that the synthesis of most lysosomal enzymes and membrane proteins is controlled by transcription factor EB (TFEB), which promotes the transcription of nuclear genes.[13][14] Mutations in the genes for these enzymes are responsible for more than 50 different human genetic disorders, which are collectively known as lysosomal storage diseases. These diseases result from an accumulation of specific substrates, due to the inability to break them down. These genetic defects are related to several neurodegenerative disorders, cancers, cardiovascular diseases, and aging-related diseases.[15][16][17]
Lysosomes should not be confused with liposomes, or with micelles.
Discovery
.jpg)
Christian de Duve, the chairman of the Laboratory of Physiological Chemistry at the Catholic University of Louvain in Belgium, had been studying the mechanism of action of a pancreatic hormone insulin in liver cells. By 1949, he and his team had focused on the enzyme called glucose 6-phosphatase, which is the first crucial enzyme in sugar metabolism and the target of insulin. They already suspected that this enzyme played a key role in regulating blood sugar levels. However, even after a series of experiments, they failed to purify and isolate the enzyme from the cellular extracts. Therefore, they tried a more arduous procedure of cell fractionation, by which cellular components are separated based on their sizes using centrifugation.
They succeeded in detecting the enzyme activity from the microsomal fraction. This was the crucial step in the serendipitous discovery of lysosomes. To estimate this enzyme activity, they used that of the standardized enzyme acid phosphatase and found that the activity was only 10% of the expected value. One day, the enzyme activity of purified cell fractions which had been refrigerated for five days was measured. Surprisingly, the enzyme activity was increased to normal of that of the fresh sample. The result was the same no matter how many times they repeated the estimation, and led to the conclusion that a membrane-like barrier limited the accessibility of the enzyme to its substrate, and that the enzymes were able to diffuse after a few days (and react with their substrate). They described this membrane-like barrier as a "saclike structure surrounded by a membrane and containing acid phosphatase."[18]
It became clear that this enzyme from the cell fraction came from membranous fractions, which were definitely cell organelles, and in 1955 De Duve named them "lysosomes" to reflect their digestive properties.[19] The same year, Alex B. Novikoff from the University of Vermont visited de Duve's laboratory, and successfully obtained the first electron micrographs of the new organelle. Using a staining method for acid phosphatase, de Duve and Novikoff confirmed the location of the hydrolytic enzymes of lysosomes using light and electron microscopic studies.[20][21] de Duve won the Nobel Prize in Physiology or Medicine in 1974 for this discovery.
Originally, De Duve had termed the organelles the "suicide bags" or "suicide sacs" of the cells, for their hypothesized role in apoptosis.[22] However, it has since been concluded that they only play a minor role in cell death.[23]
Function and structure
Lysosomes contain a variety of enzymes, enabling the cell to break down various biomolecules it engulfs, including peptides, nucleic acids, carbohydrates, and lipids (lysosomal lipase). The enzymes responsible for this hydrolysis require an acidic environment for optimal activity.
In addition to being able to break down polymers, lysosomes are capable of fusing with other organelles & digesting large structures or cellular debris; through cooperation with phagosomes, they are able to conduct autophagy, clearing out damaged structures. Similarly, they are able to break-down virus particles or bacteria in phagocytosis of macrophages.
The size of lysosomes varies from 0.1 μm to 1.2 μm.[24] With a pH ranging from ~4.5–5.0, the interior of the lysosomes is acidic compared to the slightly basic cytosol (pH 7.2). The lysosomal membrane protects the cytosol, and therefore the rest of the cell, from the degradative enzymes within the lysosome. The cell is additionally protected from any lysosomal acid hydrolases that drain into the cytosol, as these enzymes are pH-sensitive and do not function well or at all in the alkaline environment of the cytosol. This ensures that cytosolic molecules and organelles are not destroyed in case there is leakage of the hydrolytic enzymes from the lysosome.
The lysosome maintains its pH differential by pumping in protons (H+ ions) from the cytosol across the membrane via proton pumps and chloride ion channels. Vacuolar-ATPases are responsible for transport of protons, while the counter transport of chloride ions is performed by ClC-7 Cl−/H+ antiporter. In this way a steady acidic environment is maintained.[25][26]
It sources its versatile capacity for degradation by import of enzymes with specificity for different substrates; cathepsins are the major class of hydrolytic enzymes, while lysosomal alpha-glucosidase is responsible for carbohydrates, and lysosomal acid phosphatase is necessary to release phosphate groups of phospholipids.
Formation
Many components of animal cells are recycled by transferring them inside or embedded in sections of membrane. For instance, in endocytosis (more specifically, macropinocytosis), a portion of the cell's plasma membrane pinches off to form vesicles that will eventually fuse with an organelle within the cell. Without active replenishment, the plasma membrane would continuously decrease in size. It is thought that lysosomes participate in this dynamic membrane exchange system and are formed by a gradual maturation process from endosomes.[27][28]
The production of lysosomal proteins suggests one method of lysosome sustainment. Lysosomal protein genes are transcribed in the nucleus in a process that is controlled by transcription factor EB (TFEB).[14] mRNA transcripts exit the nucleus into the cytosol, where they are translated by ribosomes. The nascent peptide chains are translocated into the rough endoplasmic reticulum, where they are modified. Lysosomal soluble proteins exit the endoplasmic reticulum via COPII-coated vesicles after recruitment by the EGRESS complex (ER-to-Golgi relaying of enzymes of the lysosomal system), which is composed of CLN6 and CLN8 proteins.[9][10] COPII vesicles then deliver lysosomal enzymes to the Golgi apparatus, where a specific lysosomal tag, mannose 6-phosphate, is added to the peptides. The presence of these tags allow for binding to mannose 6-phosphate receptors in the Golgi apparatus, a phenomenon that is crucial for proper packaging into vesicles destined for the lysosomal system.[29]
Upon leaving the Golgi apparatus, the lysosomal enzyme-filled vesicle fuses with a late endosome, a relatively acidic organelle with an approximate pH of 5.5. This acidic environment causes dissociation of the lysosomal enzymes from the mannose 6-phosphate receptors. The enzymes are packed into vesicles for further transport to established lysosomes.[29] The late endosome itself can eventually grow into a mature lysosome, as evidenced by the transport of endosomal membrane components from the lysosomes back to the endosomes.[27]
Pathogen entry
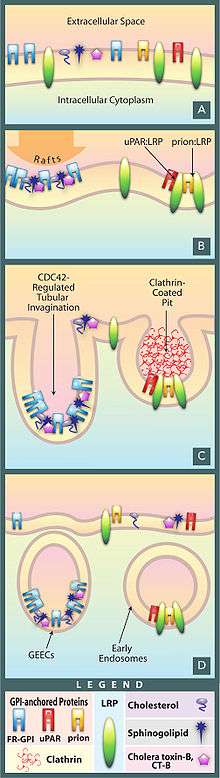
As the endpoint of endocytosis, the lysosome also acts as a safeguard in preventing pathogens from being able to reach the cytoplasm before being degraded. Pathogens often hijack endocytotic pathways such as pinocytosis in order to gain entry into the cell. The lysosome prevents easy entry into the cell by hydrolyzing the biomolecules of pathogens necessary for their replication strategies; reduced Lysosomal activity results in an increase in viral infectivity, including HIV.[30] In addition, AB5 toxins such as cholera hijack the endosomal pathway while evading lysosomal degradation.[30]
Clinical significance
Lysosomes are involved in a group of genetically inherited deficiencies, or mutations called lysosomal storage diseases (LSD), inborn errors of metabolism caused by a dysfunction of one of the enzymes. The rate of incidence is estimated to be 1 in 5,000 births, and the true figure expected to be higher as many cases are likely to be undiagnosed or misdiagnosed. The primary cause is deficiency of an acid hydrolase. Other conditions are due to defects in lysosomal membrane proteins that fail to transport the enzyme, non-enzymatic soluble lysosomal proteins. The initial effect of such disorders is accumulation of specific macromolecules or monomeric compounds inside the endosomal–autophagic–lysosomal system.[15] This results in abnormal signaling pathways, calcium homeostasis, lipid biosynthesis and degradation and intracellular trafficking, ultimately leading to pathogenetic disorders. The organs most affected are brain, viscera, bone and cartilage.[31][32]
There is no direct medical treatment to cure LSDs.[33] The most common LSD is Gaucher's disease, which is due to deficiency of the enzyme glucocerebrosidase. Consequently, the enzyme substrate, the fatty acid glucosylceramide accumulates, particularly in white blood cells, which in turn affects spleen, liver, kidneys, lungs, brain and bone marrow. The disease is characterized by bruises, fatigue, anaemia, low blood platelets, osteoporosis, and enlargement of the liver and spleen.[34][35] As of 2017, enzyme replacement therapy is available for treating 8 of the 50-60 known LDs.[36]
The most severe and rarely found, lysosomal storage disease is inclusion cell disease.[37]
Metachromatic leukodystrophy is another lysosomal storage disease that also affects sphingolipid metabolism.
Dysfunctional lysosome activity is also heavily implicated in the biology of aging, and age-related diseases such as Alzheimer's, Parkinson's, and cardiovascular disease. [38][39]
Different enzymes present in Lysosomes [40]
| Sr. No | Enzymes | Substrate |
|---|---|---|
| 1 | Phosphates | |
| A- Acid phosphatase | Most phosphomonoesters | |
| B- Acid phosphodiesterase | Oligonucleotides and phosphodiesterase | |
| 2 | Nucleases | |
| A- Acid ribonuclease | RNA | |
| B- Acid deoxyribonuclease | DNA | |
| 3 | Polysaccharides/ mucopolysaccharides hydrolyzing enzymes | |
| A- beta Galactosidase | Galactosides | |
| B- alfa Glucosidase | Glycogen | |
| C- alfa Mannosidase | Mannosides, glycoproteins | |
| D- beta Glucoronidase | Polysaccharides and mucopolyssacharides | |
| E- Lysozymes | Bacterial cell walls and mucopolyssacharides | |
| F- Hyaluronidase | Hyaluronic acids, chondroitin sulphates | |
| H- Arylsulphatase | Organic sulfates | |
| 4 | Proteases | |
| A- Cathepsin(s) | Proteins | |
| B- Collagenase | Collagen | |
| C- Peptidase | Peptides | |
| 5 | Lipid degrading enzymes | |
| A- Esterase | Fatty acyl esters | |
| B- Phospolipase | Phospholipids |
Lysosomotropism
Weak bases with lipophilic properties accumulate in acidic intracellular compartments like lysosomes. While the plasma and lysosomal membranes are permeable for neutral and uncharged species of weak bases, the charged protonated species of weak bases do not permeate biomembranes and accumulate within lysosomes. The concentration within lysosomes may reach levels 100 to 1000 fold higher than extracellular concentrations. This phenomenon is called lysosomotropism,[41] "acid trapping" or "proton pump" effect.[42] The amount of accumulation of lysosomotropic compounds may be estimated using a cell-based mathematical model.[43]
A significant part of the clinically approved drugs are lipophilic weak bases with lysosomotropic properties. This explains a number of pharmacological properties of these drugs, such as high tissue-to-blood concentration gradients or long tissue elimination half-lifes; these properties have been found for drugs such as haloperidol,[44] levomepromazine,[45] and amantadine.[46] However, high tissue concentrations and long elimination half-lives are explained also by lipophilicity and absorption of drugs to fatty tissue structures. Important lysosomal enzymes, such as acid sphingomyelinase, may be inhibited by lysosomally accumulated drugs.[47][48] Such compounds are termed FIASMAs (functional inhibitor of acid sphingomyelinase)[49] and include for example fluoxetine, sertraline, or amitriptyline.
Ambroxol is a lysosomotropic drug of clinical use to treat conditions of productive cough for its mucolytic action. Ambroxol triggers the exocytosis of lysosomes via neutralization of lysosomal pH and calcium release from acidic calcium stores.[50] Presumably for this reason, Ambroxol was also found to improve cellular function in some disease of lysosomal origin such as Parkinson's or lysosomal storage disease.[51][52]
Systemic lupus erythematosus
Impaired lysosome function is prominent in systemic lupus erythematosus preventing macrophages and monocytes from degrading neutrophil extracellular traps[53] and immune complexes.[54][55][56] The failure to degrade internalized immune complexes stems from chronic mTORC2 activity, which impairs lysosome acidification.[57] As a result, immune complexes in the lysosome recycle to the surface of macrophages causing an accumulation of nuclear antigens upstream of multiple lupus-associated pathologies.[54][58][59]
Controversy in botany
By scientific convention, the term lysosome is applied to these vesicular organelles only in animals, and the term vacuole is applied to those in plants, fungi and algae (some animal cells also have vacuoles). Discoveries in plant cells since the 1970s started to challenge this definition. Plant vacuoles are found to be much more diverse in structure and function than previously thought.[60][61] Some vacuoles contain their own hydrolytic enzymes and perform the classic lysosomal activity, which is autophagy.[62][63][64] These vacuoles are therefore seen as fulfilling the role of the animal lysosome. Based on de Duve's description that "only when considered as part of a system involved directly or indirectly in intracellular digestion does the term lysosome describe a physiological unit", some botanists strongly argued that these vacuoles are lysosomes.[65] However, this is not universally accepted as the vacuoles are strictly not similar to lysosomes, such as in their specific enzymes and lack of phagocytic functions.[66] Vacuoles do not have catabolic activity and do not undergo exocytosis as lysosomes do.[67]
Etymology and pronunciation
The word lysosome (/ˈlaɪsoʊsoʊm/, /ˈlaɪzəzoʊm/) is New Latin that uses the combining forms lyso- (referring to lysis and derived from the Latin lysis, meaning "to loosen", via Ancient Greek λύσις [lúsis]), and -some, from soma, "body", yielding "body that lyses" or "lytic body". The adjectival form is lysosomal. The forms *lyosome and *lyosomal are much rarer; they use the lyo- form of the prefix but are often treated by readers and editors as mere unthinking replications of typos, which has no doubt been true as often as not.
References
- By convention similar cells in plants are called vacuoles, see #Controversy in botany
- Ohkuma S, Poole B (July 1978). "Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents". Proceedings of the National Academy of Sciences of the United States of America. 75 (7): 3327–31. doi:10.1073/pnas.75.7.3327. PMC 392768. PMID 28524.
- Settembre C, Fraldi A, Medina DL, Ballabio A (May 2013). "Signals from the lysosome: a control centre for cellular clearance and energy metabolism". Nature Reviews Molecular Cell Biology. 14 (5): 283–96. doi:10.1038/nrm3565. PMC 4387238. PMID 23609508.
- Holtzclaw FW, et al. (2008). AP* Biology: to Accompany Biology (8th AP ed.). Pearson Benjamin Cummings.
- Underwood, Emily (2018). "When the brain's waste disposal system fails". Knowable Magazine. doi:10.1146/knowable-121118-1.
- Lüllmznn-Rauch R (2005). "History and Morphology of Lysosome". In Zaftig P (ed.). Lysosomes (Online-Ausg. 1 ed.). Georgetown, Tex.: Landes Bioscience/Eurekah.com. pp. 1–16. ISBN 978-0-387-28957-1.
- Xu H, Ren D (2015). "Lysosomal physiology". Annual Review of Physiology. 77 (1): 57–80. doi:10.1146/annurev-physiol-021014-071649. PMC 4524569. PMID 25668017.
- "Lysosomal Enzymes". www.rndsystems.com. R&D Systems. Retrieved 4 October 2016.
- di Ronza A, Bajaj L, Sharma J, Sanagasetti D, Lotfi P, Adamski CJ, Collette J, Palmieri M, Amawi A, Popp L, Chang KT, Meschini MC, Leung HE, Segatori L, Simonati A, Sifers RN, Santorelli FM, Sardiello M (December 2018). "CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis". Nature Cell Biology. 20 (12): 1370–1377. doi:10.1038/s41556-018-0228-7. PMC 6277210. PMID 30397314.
- Bajaj L, Sharma J, di Ronza A, Zhang P, Eblimit A, Pal R, Roman D, Collette JR, Booth C, Chang KT, Sifers RN, Jung SY, Weimer JM, Chen R, Schekman RW, Sardiello M (June 2020). "A CLN6-CLN8 complex recruits lysosomal enzymes at the ER for Golgi transfer". J Clin Invest. doi:10.1172/JCI130955. PMID 32597833.
- Saftig P, Klumperman J (September 2009). "Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function". Nature Reviews Molecular Cell Biology. 10 (9): 623–35. doi:10.1038/nrm2745. PMID 19672277.
- Samie MA, Xu H (June 2014). "Lysosomal exocytosis and lipid storage disorders". Journal of Lipid Research. 55 (6): 995–1009. doi:10.1194/jlr.R046896. PMC 4031951. PMID 24668941.
- Underwood, Emily (2018). "When the brain's waste disposal system fails". Knowable Magazine. doi:10.1146/knowable-121118-1.
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A (July 2009). "A gene network regulating lysosomal biogenesis and function". Science. 325 (5939): 473–7. Bibcode:2009Sci...325..473S. doi:10.1126/science.1174447. PMID 19556463.
- Platt FM, Boland B, van der Spoel AC (November 2012). "The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction". The Journal of Cell Biology. 199 (5): 723–34. doi:10.1083/jcb.201208152. PMC 3514785. PMID 23185029.
- He LQ, Lu JH, Yue ZY (May 2013). "Autophagy in ageing and ageing-associated diseases". Acta Pharmacologica Sinica. 34 (5): 605–11. doi:10.1038/aps.2012.188. PMC 3647216. PMID 23416930.
- Carmona-Gutierrez, Didac; Hughes, Adam L.; Madeo, Frank; Ruckenstuhl, Christoph (1 December 2016). "The crucial impact of lysosomes in aging and longevity". Ageing Research Reviews. Lysosomes in Aging. 32: 2–12. doi:10.1016/j.arr.2016.04.009. ISSN 1568-1637. PMC 5081277. PMID 27125853.
- Susana Castro-Obregon (2010). "The Discovery of Lysosomes and Autophagy". Nature Education. 3 (9): 49.
- de Duve C (September 2005). "The lysosome turns fifty". Nature Cell Biology. 7 (9): 847–9. doi:10.1038/ncb0905-847. PMID 16136179.
- Novikoff AB, Beaufay H, De Duve C (July 1956). "Electron microscopy of lysosomerich fractions from rat liver". The Journal of Biophysical and Biochemical Cytology. 2 (4 Suppl): 179–84. doi:10.1083/jcb.2.4.179. PMC 2229688. PMID 13357540.
- Klionsky DJ (August 2008). "Autophagy revisited: a conversation with Christian de Duve". Autophagy. 4 (6): 740–3. doi:10.4161/auto.6398. PMID 18567941.
- Hayashi, Teru, and others. "Subcellular Particles." Subcellular Particles., 1959.
- Turk B, Turk V (2009). "Lysosomes as 'Suicide Bags' in Cell Death: Myth or Reality?". The Journal of Biological Chemistry. 284 (33): 21783–87. doi:10.1074/jbc.R109.023820. PMC 2755904. PMID 19473965.
- Kuehnel W (2003). Color Atlas of Cytology, Histology, & Microscopic Anatomy (4th ed.). Thieme. p. 34. ISBN 978-1-58890-175-0.
- Mindell JA (2012). "Lysosomal acidification mechanisms". Annual Review of Physiology. 74 (1): 69–86. doi:10.1146/annurev-physiol-012110-142317. PMID 22335796.
- Ishida Y, Nayak S, Mindell JA, Grabe M (June 2013). "A model of lysosomal pH regulation". The Journal of General Physiology. 141 (6): 705–20. doi:10.1085/jgp.201210930. PMC 3664703. PMID 23712550.
- Alberts B, et al. (2002). Molecular biology of the cell (4th ed.). New York: Garland Science. ISBN 978-0-8153-3218-3.
- Falcone S, Cocucci E, Podini P, Kirchhausen T, Clementi E, Meldolesi J (November 2006). "Macropinocytosis: regulated coordination of endocytic and exocytic membrane traffic events". Journal of Cell Science. 119 (Pt 22): 4758–69. doi:10.1242/jcs.03238. PMID 17077125.
- Lodish H, et al. (2000). Molecular cell biology (4th ed.). New York: Scientific American Books. ISBN 978-0-7167-3136-8.
- Wei BL, Denton PW, O'Neill E, Luo T, Foster JL, Garcia JV (May 2005). "Inhibition of lysosome and proteasome function enhances human immunodeficiency virus type 1 infection". Journal of Virology. 79 (9): 5705–12. doi:10.1128/jvi.79.9.5705-5712.2005. PMC 1082736. PMID 15827185.
- Schultz ML, Tecedor L, Chang M, Davidson BL (August 2011). "Clarifying lysosomal storage diseases". Trends in Neurosciences. 34 (8): 401–10. doi:10.1016/j.tins.2011.05.006. PMC 3153126. PMID 21723623.
- Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A (May 2012). "Autophagy in lysosomal storage disorders". Autophagy. 8 (5): 719–30. doi:10.4161/auto.19469. PMC 3378416. PMID 22647656.
- .Parenti G, Pignata C, Vajro P, Salerno M (January 2013). "New strategies for the treatment of lysosomal storage diseases (review)". International Journal of Molecular Medicine. 31 (1): 11–20. doi:10.3892/ijmm.2012.1187. PMID 23165354.
- Rosenbloom BE, Weinreb NJ (2013). "Gaucher disease: a comprehensive review". Critical Reviews in Oncogenesis. 18 (3): 163–75. doi:10.1615/CritRevOncog.2013006060. PMID 23510062.
- Sidransky E (October 2012). "Gaucher disease: insights from a rare Mendelian disorder". Discovery Medicine. 14 (77): 273–81. PMC 4141347. PMID 23114583.
- Solomon M, Muro S (September 2017). "Lysosomal enzyme replacement therapies: Historical development, clinical outcomes, and future perspectives". Advanced Drug Delivery Reviews. 118: 109–134. doi:10.1016/j.addr.2017.05.004. PMC 5828774. PMID 28502768.
- Alberts, Bruce (2002). Molecular biology of the cell (4th ed.). Garland Science. p. 744. ISBN 978-0815340720.
- Carmona-Gutierrez, Didac; Hughes, Adam L.; Madeo, Frank; Ruckenstuhl, Christoph (1 December 2016). "The crucial impact of lysosomes in aging and longevity". Ageing Research Reviews. Lysosomes in Aging. 32: 2–12. doi:10.1016/j.arr.2016.04.009. ISSN 1568-1637. PMC 5081277. PMID 27125853.
- Finkbeiner, Steven (1 April 2019). "The Autophagy Lysosomal Pathway and Neurodegeneration". Cold Spring Harbor Perspectives in Biology: a033993. doi:10.1101/cshperspect.a033993. ISSN 1943-0264. PMC 6773515. PMID 30936119.
- Pranav Kumar. (2013). Life Sciences : Fundamentals and practice. Mina, Usha. (3rd ed.). New Delhi: Pathfinder Academy. ISBN 978-81-906427-0-5. OCLC 857764171.
- de Duve C, de Barsy T, Poole B, Trouet A, Tulkens P, Van Hoof F (September 1974). "Commentary. Lysosomotropic agents". Biochemical Pharmacology. 23 (18): 2495–531. doi:10.1016/0006-2952(74)90174-9. PMID 4606365.
- Traganos F, Darzynkiewicz Z (1994). "Lysosomal proton pump activity: supravital cell staining with acridine orange differentiates leukocyte subpopulations". Methods Cell Biol. 41: 185–94. doi:10.1016/S0091-679X(08)61717-3. PMID 7532261.
- Trapp S, Rosania GR, Horobin RW, Kornhuber J (October 2008). "Quantitative modeling of selective lysosomal targeting for drug design". European Biophysics Journal. 37 (8): 1317–28. doi:10.1007/s00249-008-0338-4. PMC 2711917. PMID 18504571.
- Kornhuber J, Schultz A, Wiltfang J, Meineke I, Gleiter CH, Zöchling R, Boissl KW, Leblhuber F, Riederer P (June 1999). "Persistence of haloperidol in human brain tissue". The American Journal of Psychiatry. 156 (6): 885–90. doi:10.1176/ajp.156.6.885. PMID 10360127.
- Kornhuber J, Weigmann H, Röhrich J, Wiltfang J, Bleich S, Meineke I, Zöchling R, Härtter S, Riederer P, Hiemke C (March 2006). "Region specific distribution of levomepromazine in the human brain". Journal of Neural Transmission. 113 (3): 387–97. doi:10.1007/s00702-005-0331-3. PMID 15997416.
- Kornhuber J, Quack G, Danysz W, Jellinger K, Danielczyk W, Gsell W, Riederer P (July 1995). "Therapeutic brain concentration of the NMDA receptor antagonist amantadine". Neuropharmacology. 34 (7): 713–21. doi:10.1016/0028-3908(95)00056-c. PMID 8532138.
- Kornhuber J, Tripal P, Reichel M, Terfloth L, Bleich S, Wiltfang J, Gulbins E (January 2008). "Identification of new functional inhibitors of acid sphingomyelinase using a structure-property-activity relation model". Journal of Medicinal Chemistry. 51 (2): 219–37. CiteSeerX 10.1.1.324.8854. doi:10.1021/jm070524a. PMID 18027916.
- Kornhuber J, Muehlbacher M, Trapp S, Pechmann S, Friedl A, Reichel M, Mühle C, Terfloth L, Groemer TW, Spitzer GM, Liedl KR, Gulbins E, Tripal P (2011). Riezman H (ed.). "Identification of novel functional inhibitors of acid sphingomyelinase". PLOS ONE. 6 (8): e23852. doi:10.1371/journal.pone.0023852. PMC 3166082. PMID 21909365.
- Kornhuber J, Tripal P, Reichel M, Mühle C, Rhein C, Muehlbacher M, Groemer TW, Gulbins E (2010). "Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications". Cellular Physiology and Biochemistry. 26 (1): 9–20. doi:10.1159/000315101. PMID 20502000.
- Fois G, Hobi N, Felder E, Ziegler A, Miklavc P, Walther P, Radermacher P, Haller T, Dietl P (December 2015). "A new role for an old drug: Ambroxol triggers lysosomal exocytosis via pH-dependent Ca²⁺ release from acidic Ca²⁺ stores". Cell Calcium. 58 (6): 628–37. doi:10.1016/j.ceca.2015.10.002. PMID 26560688.
- Albin RL, Dauer WT (May 2014). "Magic shotgun for Parkinson's disease?". Brain. 137 (Pt 5): 1274–5. doi:10.1093/brain/awu076. PMID 24771397.
- McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, Foltynie T, Cooper JM, Abramov AY, Gegg M, Schapira AH (May 2014). "Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells". Brain. 137 (Pt 5): 1481–95. doi:10.1093/brain/awu020. PMC 3999713. PMID 24574503.
- Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A (May 2010). "Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis". Proceedings of the National Academy of Sciences of the United States of America. 107 (21): 9813–8. doi:10.1073/pnas.0909927107. PMC 2906830. PMID 20439745.
- Monteith AJ, Kang S, Scott E, Hillman K, Rajfur Z, Jacobson K, Costello MJ, Vilen BJ (April 2016). "Defects in lysosomal maturation facilitate the activation of innate sensors in systemic lupus erythematosus". Proceedings of the National Academy of Sciences of the United States of America. 113 (15): E2142–51. doi:10.1073/pnas.1513943113. PMC 4839468. PMID 27035940.
- Kavai M, Szegedi G (August 2007). "Immune complex clearance by monocytes and macrophages in systemic lupus erythematosus". Autoimmunity Reviews. 6 (7): 497–502. doi:10.1016/j.autrev.2007.01.017. PMID 17643939.
- Kávai M, Csipö I, Sonkoly I, Csongor J, Szegedi GY (November 1986). "Defective immune complex degradation by monocytes in patients with systemic lupus erythematosus". Scandinavian Journal of Immunology. 24 (5): 527–32. doi:10.1111/j.1365-3083.1986.tb02167.x. PMID 3787186.
- Monteith AJ, Vincent HA, Kang S, Li P, Claiborne TM, Rajfur Z, Jacobson K, Moorman NJ, Vilen BJ (July 2018). "mTORC2 Activity Disrupts Lysosome Acidification in Systemic Lupus Erythematosus by Impairing Caspase-1 Cleavage of Rab39a". Journal of Immunology. 201 (2): 371–382. doi:10.4049/jimmunol.1701712. PMC 6039264. PMID 29866702.
- Kang S, Rogers JL, Monteith AJ, Jiang C, Schmitz J, Clarke SH, Tarrant TK, Truong YK, Diaz M, Fedoriw Y, Vilen BJ (May 2016). "Apoptotic Debris Accumulates on Hematopoietic Cells and Promotes Disease in Murine and Human Systemic Lupus Erythematosus". Journal of Immunology. 196 (10): 4030–9. doi:10.4049/jimmunol.1500418. PMC 4868781. PMID 27059595.
- Kang S, Fedoriw Y, Brenneman EK, Truong YK, Kikly K, Vilen BJ (April 2017). "BAFF Induces Tertiary Lymphoid Structures and Positions T Cells within the Glomeruli during Lupus Nephritis". Journal of Immunology. 198 (7): 2602–2611. doi:10.4049/jimmunol.1600281. PMC 5360485. PMID 28235864.
- Marty F (April 1999). "Plant vacuoles". The Plant Cell. 11 (4): 587–600. doi:10.2307/3870886. JSTOR 3870886. PMC 144210. PMID 10213780.
- Samaj J, Read ND, Volkmann D, Menzel D, Baluska F (August 2005). "The endocytic network in plants". Trends in Cell Biology. 15 (8): 425–33. doi:10.1016/j.tcb.2005.06.006. PMID 16006126.
- Matile, P (1978). "Biochemistry and Function of Vacuoles". Annual Review of Plant Physiology. 29 (1): 193–213. doi:10.1146/annurev.pp.29.060178.001205.
- Moriyasu Y, Ohsumi Y (August 1996). "Autophagy in Tobacco Suspension-Cultured Cells in Response to Sucrose Starvation". Plant Physiology. 111 (4): 1233–1241. doi:10.1104/pp.111.4.1233. PMC 161001. PMID 12226358.
- Jiao BB, Wang JJ, Zhu XD, Zeng LJ, Li Q, He ZH (January 2012). "A novel protein RLS1 with NB-ARM domains is involved in chloroplast degradation during leaf senescence in rice". Molecular Plant. 5 (1): 205–17. doi:10.1093/mp/ssr081. PMID 21980143.
- Swanson SJ, Bethke PC, Jones RL (May 1998). "Barley aleurone cells contain two types of vacuoles. Characterization Of lytic organelles by use of fluorescent probes". The Plant Cell. 10 (5): 685–98. doi:10.2307/3870657. JSTOR 3870657. PMC 144374. PMID 9596630.
- Holtzman E (1989). Lysosomes. New York: Plenum Press. pp. 7, 15. ISBN 978-0306-4-3126-5.
- De DN (2000). Plant Cell Vacuoles: An Introduction. Australia: Csiro Publishing. ISBN 978-0-643-09944-9.
External links
| Look up lysosome in Wiktionary, the free dictionary. |

- 3D structures of proteins associated with lysosome membrane
- Hide and Seek Foundation For Lysosomal Research
- Lysosomal Disease Network, a research consortium funded by the NIH through its NCATS/Rare Diseases Clinical Research Network
- Self-Destructive Behavior in Cells May Hold Key to a Longer Life
- Mutations in the Lysosomal Enzyme–Targeting Pathway and Persistent Stuttering
- Animation showing how lysosomes are made, and their function