Heteroplasmy
Heteroplasmy is the presence of more than one type of organellar genome (mitochondrial DNA or plastid DNA) within a cell or individual. It is an important factor in considering the severity of mitochondrial diseases. Because most eukaryotic cells contain many hundreds of mitochondria with hundreds of copies of mitochondrial DNA, it is common for mutations to affect only some mitochondria, leaving most unaffected.
Although detrimental scenarios are well-studied, heteroplasmy can also be beneficial. For example, centenarians show a higher than average degree of heteroplasmy.[1]
Microheteroplasmy is present in most individuals. This refers to hundreds of independent mutations in one organism, with each mutation found in about 1–2% of all mitochondrial genomes.[2]
Types of heteroplasmy
In order for heteroplasmy to occur, organelles must contain a genome and, in turn, a genotype. In animals, mitochondria are the only organelles that contain their own genomes, so these organisms will only have mitochondrial heteroplasmy. In contrast, photosynthetic plants contain mitochondria and chloroplasts, each of which contains plastid genomes. Therefore, plant heteroplasmy occurs in two dimensions.[3]
Organelle inheritance patterns
In 1909, while studying chloroplast genomes, Erwin Baur made the first observations about organelle inheritance patterns. Organelle genome inheritance differs from nuclear genome, and this is illustrated by four violations of Mendel's laws.[4]
- During asexual reproduction, nuclear genes never segregate during cellular divisions. This is to ensure that each daughter cell gets a copy of every gene. However, organelle genes in heteroplasmic cells can segregate because they each have several copies of their genome. This may result in daughter cells with differential proportions of organelle genotypes.[4]
- Mendel states that nuclear alleles always segregate during meiosis. However, organelle alleles may or may not do this.[4]
- Nuclear genes are inherited from a combination of alleles from both parents, making inheritance biparental. Conversely, organelle inheritance is uniparental, meaning the genes are all inherited from one parent.[4]
- It is also unlikely for organelle alleles to segregate independently, like nuclear alleles do, because plastid genes are usually on a single chromosome and recombination is limited by uniparental inheritance.[4]
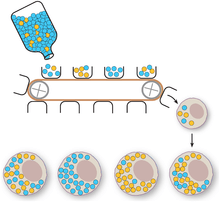
Vegetative segregation
Vegetative segregation, the random partitioning of cytoplasm, is a distinguishable characteristic of organelle heredity. During cell division, the organelles are divided equally, providing each daughter cell with a random selection of plasmid genotypes.[4]
Uniparental inheritance
Uniparental inheritance refers to the fact that, in most organisms, many offspring inherit organelle genes from only one parent. However, this is not a general law. Many organisms that have the ability to differentiate maternal and paternal sexes will produce offspring with a mixture of maternal, paternal, and biparental mitochondrial DNA.[4]
Mitochondrial bottleneck
Entities undergoing uniparental inheritance and with little to no recombination may be expected to be subject to Muller's ratchet, the inexorable accumulation of deleterious mutations until functionality is lost. Animal populations of mitochondria avoid this buildup through a developmental process known as the mtDNA bottleneck. The bottleneck exploits stochastic processes in the cell to increase in the cell-to-cell variability in mutant load as an organism develops: a single egg cell with some proportion of mutant mtDNA thus produces an embryo where different cells have different mutant loads. Cell-level selection may then act to remove those cells with more mutant mtDNA, leading to a stabilisation or reduction in mutant load between generations. The mechanism underlying the bottleneck is debated,[6][7][8] with a recent mathematical and experimental metastudy providing evidence for a combination of random partitioning of mtDNAs at cell divisions and random turnover of mtDNA molecules within the cell.[9]
The mitochondrial bottleneck concept refers to the classic evolutionary term, which is used to explain an event that reduces and specifies a population. It was developed to describe why mitochondrial DNA in an embryo might be drastically different from that of its mother. When a large population of DNA is subsampled, each sample population will receive a slightly different proportion of mitochondrial genotypes. Consequently, when paired with a high degree of replication, a rare or mutated allele can begin to proportionally dominate. In theory, this makes possible a single-generation shift of overall mitochondrial genotype.[5]
Selection
Although it is not well characterized, selection can occur for organelle genomes in heteroplasmic cells. Intracellular ("within cells") selection occurs within individual cells. It refers to the selective segregation of certain genotypes in mitochondrial DNA that allows the favoured genotype to thrive. Intercellular ("between cells") selection occurs on a larger scale, and refers to the preferential growth of cells that have greater numbers of a certain mitochondrial genotype.[4] Selective differences can occur between naturally occurring, non-pathological mtDNA types when mixed in cells, and may depend on tissue type, age, and genetic distance.[10] Selective differences between naturally occurring mtDNA types may pose challenges for gene therapies.[11]
In mitochondrial DNA, there is evidence for potent germline purifying selection, as well as purifying selection during embryogenesis. Additionally, there is a dose-dependent decrease in reproduction ability for females that have mutations in mitochondrial DNA. This demonstrates another selection mechanism to prevent the evolutionary preservation of harmful mutations.[5]
Reduced recombination
It is very rare for organelle genes from different lineages to recombine. These genomes are usually inherited uniparentally, which does not provide a recombination opportunity. If they are inherited biparentally, it is unlikely that the organelles from the parents will fuse, meaning they will not share genomes.
However, it is possible for organelle genes from the same lineage to recombine. Intramolecular and intermolecular recombination can cause inversions and repeats in chloroplast DNA, and can produce subgenomic circles in mitochondrial DNA.[4]
Mitochondrial mutations in disease
Mutations in mitochondrial DNA are usually single nucleotide substitutions, single base insertions, or deletions.
Because each cell contains thousands of mitochondria, nearly all organisms house low levels of mitochondrial variants, conferring some degree of heteroplasmy. Although a single mutational event might be rare in its generation, repeated mitotic segregation and clonal expansion can enable it to dominate the mitochondrial DNA pool over time. When this occurs, it is known as reaching threshold, and it usually results in physiological consequences.[5]
Severity and time to presentation
Symptoms of severe heteroplasmic mitochondrial disorders do not usually appear until adulthood. Many cell divisions and a great deal of time are required for a cell to accumulate enough mutant mitochondria to cause symptoms. An example of this phenomenon is Leber optic atrophy. Generally, individuals with this condition do not experience vision difficulties until they have reached adulthood. Another example is MERRF syndrome (or Myoclonic Epilepsy with Ragged Red Fibers). In MELAS, heteroplasmy explains the variation in severity of the disease among siblings.
Screening
Preimplantation genetic screening (PGS) can be used to quantitate the risk of a child of being affected by a mitochondrial disease. In most cases, a muscle mutation level of approximately 18% or less confers a 95% risk reduction.[12]
Notable cases
One notable example of an otherwise healthy individual whose heteroplasmy was discovered incidentally is Nicholas II of Russia, whose heteroplasmy (and that of his brother) served to convince Russian authorities of the authenticity of his remains.[14]
See also
- Homoplasmy
- Microheteroplasmy
- Mitochondrial diseases
Notes and references
- Rose G, Passarino G, Scornaienchi V, Romeo G, Dato S, Bellizzi D, Mari V, Feraco E, Maletta R, Bruni A, Franceschi C, De Benedictis G (2007). "The mitochondrial DNA control region shows genetically correlated levels of heteroplasmy in leukocytes of centenarians and their offspring". BMC Genomics. 8: 293. doi:10.1186/1471-2164-8-293. PMC 2014781. PMID 17727699.
- Smigrodzki, R. M.; Khan, S. M. (2005). "Mitochondrial Microheteroplasmy and a Theory of Aging and Age-Related Disease". Rejuvenation Research. 8 (3): 172–198. doi:10.1089/rej.2005.8.172. PMID 16144471.
- Korpelainen, H. (2004). "The evolutionary processes of mitochondrial and chloroplast genomes differ from those of nuclear genomes". Die Naturwissenschaften. 91 (11): 505–518. doi:10.1007/s00114-004-0571-3. PMID 15452701.
- Birky, C. William (2001). "The Inheritance of Genes in Mitochondria and Chloroplasts: Laws, Mechanisms, and Models". Annu. Rev. Genet. 35: 125–148. doi:10.1146/annurev.genet.35.102401.090231. PMID 11700280.
- Stewart, J., Larsson, N. (2014). "Keeping mtDNA in shape between generations". PLOS Genetics. 10 (10): e1004670. doi:10.1371/journal.pgen.1004670. PMC 4191934. PMID 25299061.CS1 maint: multiple names: authors list (link)
- Cree, L.M., Samuels, D.C., de Sousa Lopes, S.C., Rajasimha, H.K., Wonnapinij, P., Mann, J.R., Dahl, H.H.M. and Chinnery, P.F. (2008). "A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes". Nature Genetics. 40 (2): 249–254. doi:10.1038/ng.2007.63. PMID 18223651.CS1 maint: multiple names: authors list (link)
- Cao, L., Shitara, H., Horii, T., Nagao, Y., Imai, H., Abe, K., Hara, T., Hayashi, J.I. and Yonekawa, H. (2007). "The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells". Nature Genetics. 39 (3): 386–390. doi:10.1038/ng1970. PMID 17293866.CS1 maint: multiple names: authors list (link)
- Wai, T., Teoli, D. and Shoubridge, E.A. (2008). "The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes". Nature Genetics. 40 (12): 1484–1488. doi:10.1038/ng.258. PMID 19029901.CS1 maint: multiple names: authors list (link)
- Johnston, I.G., Burgstaller, J.P., Havlicek, V., Kolbe, T., Rülicke, T., Brem, G., Poulton, J. and Jones, N.S. (2015). "Stochastic modelling, Bayesian inference, and new in vivo measurements elucidate the debated mtDNA bottleneck mechanism". eLife. 4: e07464. doi:10.7554/eLife.07464. PMC 4486817. PMID 26035426.CS1 maint: multiple names: authors list (link)
- Burgstaller, J.P., Johnston, I.G., Jones, N.S., Albrechtová, J., Kolbe, T., Vogl, C., Futschik, A., Mayrhofer, C., Klein, D., Sabitzer, S. and Blattner, M. (2014). "mtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage". Cell Reports. 7 (6): 2031–2041. doi:10.1016/j.celrep.2014.05.020. PMC 4570183. PMID 24910436.CS1 maint: multiple names: authors list (link)
- Burgstaller, J.P., Johnston, I.G. and Poulton, J. (2015). "Mitochondrial DNA disease and developmental implications for reproductive strategies". Molecular Human Reproduction. 21 (1): 11–22. doi:10.1093/molehr/gau090. PMC 4275042. PMID 25425607.CS1 maint: multiple names: authors list (link)
- Hellebrekers, D. M. E. I.; Wolfe, R.; Hendrickx, A. T. M.; De Coo, I. F. M.; De Die, C. E.; Geraedts, J. P. M.; Chinnery, P. F.; Smeets, H. J. M. (2012). "PGD and heteroplasmic mitochondrial DNA point mutations: A systematic review estimating the chance of healthy offspring". Human Reproduction Update. 18 (4): 341–349. doi:10.1093/humupd/dms008. PMID 22456975.
- Coble MD, Loreille OM, Wadhams MJ, Edson SM, Maynard K, Meyer CE, Niederstätter H, Berger C, Berger B, Falsetti AB, Gill P, Parson W, Finelli LN (2009). "Mystery solved: the identification of the two missing Romanov children using DNA analysis". PLoS ONE. 4 (3): e4838. doi:10.1371/journal.pone.0004838. PMC 2652717. PMID 19277206.
- Ivanov PL, Wadhams MJ, Roby RK, Holland MM, Weedn VW, Parsons TJ (April 1996). "Mitochondrial DNA sequence heteroplasmy in the Grand Duke of Russia Georgij Romanov establishes the authenticity of the remains of Tsar Nicholas II". Nat. Genet. 12 (4): 417–20. doi:10.1038/ng0496-417. PMID 8630496.