Haploinsufficiency of A20
Haploinsufficiency of A20 is a rare disease caused by mutations in the gene TNFAIP3.[1][2] This gene is also known as A20.
| Haploinsufficiency of A20 | |
|---|---|
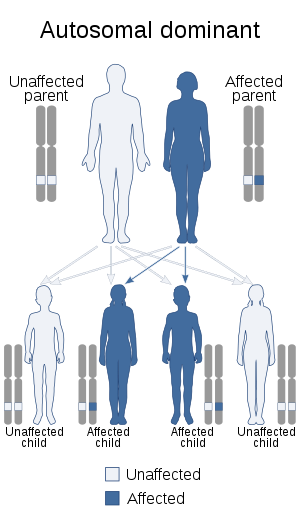 | |
| This condition is inherited in an autosomal dominant manner. | |
| Specialty | Medical genetics |
Signs and symptoms
These are variable even within families. the main features are recurrent oral, genital and/or gastrointestinal ulcers, musculoskeletal and gastrointestinal complaints, cutaneous lesions, episodic fever and recurrent infections. The age on onset is also variable ranging from the first week of life to 29 years.[3] The male:female ratio is 1:3.
Genetics
The TNFAIP3 gene is located on the long arm of chromosome 6 (6q23.3). Inheritance appears to be autosomal dominant with variable penetrance.
It appears that two copies of this gene are required to avoid inflammatory features developing - hence the name haploinsufficiency.
Pathogenesis
The gene encodes a protein - Tumor necrosis factor alpha-induced protein 3 - which inhibits the pro inflammatory actions of NF-κB. The protein encoded has both ubiquitin ligation deubiquitinase activities and forms part of the ubiquitin editing protein complex. It is involved in several biochemical pathways the details of which are still under investigation.
Diagnosis
Diagnosis is made by sequencing the TNFAIP3 gene. The usual laboratory tests are consistent with non-specific inflammation. Antinuclear antibodies and anti-dsDNA antibodies may be positive. Biopsies show non specific inflammatory changes.
Differential diagnosis
The main differential diagnosis are Behçet's disease and systemic lupus erythematosus.
Treatment
Response to colchicine has been variable. Cytokine inhibitors - including the anti-IL6 receptor biologic tocilizumab - appear to be the most effective.
Given the rarity of this condition, optimal management has not yet been definitely identified.
History
This condition was first described in 2016.[4]
References
- Demir S, Sonmez HE, OZen S (2018) Vasculitis: Decade in Review. Curr Rheumatol Rev doi: 10.2174/1573397114666180726093731
- Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P, Leavis HL, Ozen S, Schwartz DM, Stone DL, van Royen-Kerkof A, Kastner DL, Aksentijevich I, Laxer RM (2018) A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis 77(5):728-735 doi: 10.1136/annrheumdis-2017-212403
- Zheng C, Huang Y, Ye Z, Wang Y, Tang Z, Lu J, Wu J, Zhou Y, Wang L, Huang Z, Yang H, Xue A (2018) Infantile onset intractable inflammatory bowel disease due to novel heterozygous mutations in TNFAIP3 (A20). Inflamm Bowel Dis doi: 10.1093/ibd/izy165
- Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, Yang D, Demirkaya E, Takeuchi M, Tsai WL, Lyons JJ3, Yu X, Ouyang C, Chen C, Chin DT, Zaal K, Chandrasekharappa SC, P Hanson E, Yu Z, Mullikin JC, Hasni SA, Wertz IE, Ombrello AK, Stone DL, Hoffmann P, Jones A, Barham BK, Leavis HL, van Royen-Kerkof A, Sibley C, Batu ED, Gül A, Siegel RM, Boehm M, Milner JD, Ozen S, Gadina M, Chae J, Laxer RM, Kastner DL, Aksentijevich I (2016) Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet 48(1):67-73 doi: 10.1038/ng.3459
| Classification |
|---|