G2-M DNA damage checkpoint
The G2-M DNA damage checkpoint is an important cell cycle checkpoint in eukaryotic organisms that ensures that cells don't initiate mitosis until damaged or incompletely replicated DNA is sufficiently repaired. Cells which have a defective G2-M checkpoint, if they enter M phase before repairing their DNA, it leads to apoptosis or death after cell division.[1] The defining biochemical feature of this checkpoint is the activation of M-phase cyclin-CDK complexes, which phosphorylate proteins that promote spindle assembly and bring the cell to metaphase.[2]
Cyclin B-CDK 1 Activity
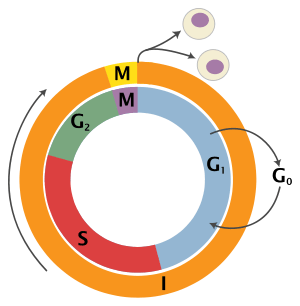
The cell cycle is driven by proteins called cyclin dependent kinases that associate with cyclin regulatory proteins at different checkpoints of the cell cycle. Different phases of the cell cycle experience activation and/or deactivation of specific cyclin-CDK complexes.
CyclinB-CDK1 activity is specific to the G2/M checkpoint. Accumulation of cyclin B increases the activity of the cyclin dependent kinase Cdk1 human homolog Cdc2 as cells prepare to enter mitosis. Cdc2 activity is further regulated by phosphorylation/dephosphorylation of its corresponding activators and inhibitors. Through a positive feedback loop, CyclinB-Cdc2 activates the phosphatase Cdc25 which in turn deactivates the CyclinB-Cdc2 inhibitors, Wee1 and Myt1. Cdc25 activates the complex through the removal of phosphates from the active site while Wee1 inactivates the complex through the phosphorylation of tyrosine residues, specifically tyrosine-15.[3]
This loop is further amplified indirectly through the coordinated interaction of the Aurora A kinase and the Bora cofactor. During the G2 phase, Bora accumulates and forms an activation complex with Aurora A. This complex then regulates the activation of Polo-like kinase 1 (Plk1). Plk1 phosphorylates Wee1, targeting it for degradation through the SCF ubiquitin ligase complex (SCF complex), and activates Cdc25 through phosphorylation with combined action activating Cdc2. The combined activity and complex of Cdc2, Cdc25, and Plk1 with the accumulation of cyclin B activates the CyclinB-Cdc2 complex, promoting entry into mitosis.[4]
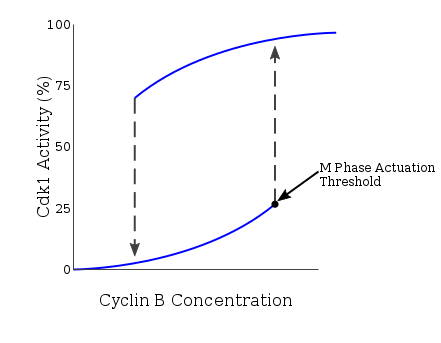
Many proteins involved in this positive feedback loop drive the activation of the CyclinB-Cdc2 complex because entry into mitosis requires an all-or-none response. The Novak-Tyson model is a mathematical model used to explain such regulatory loop that predicted the irreversible transition into mitosis driven by hysteresis.[5] Through experiments in Xenopus laevis cell-free egg extracts, such model was confirmed as the basis for entry into mitosis. Once cyclin concentration reaches a certain minimum activation threshold, Cdc2 is rapidly activated. It remains in this state until activity falls below a separate inactivation threshold at which it is abruptly inactivated through tyrosine phosphorylation by Wee1 and Myt1. In the case of unreplicated DNA, the cyclin concentration threshold for Cdc2 activation is further increased. Through this mechanism, there exists two separate steady-state conditions separated by an unstable steady state. The bistable and hysteretic nature of CyclinB-Cdc2 ensures a highly regulated nature of the G2/M checkpoint.[6]
Pathway Response to DNA Damage
Proteins that localize to sites of DNA damage in the G2 phase initiate a signaling cascade that regulates important components of the pathway, as described above, therefore controlling mitotic entry via CyclinB-Cdc2 activity. Negative regulation of CyclinB-Cdc2 activity results in a delay in mitotic entry, which is important for cells to repair any DNA damage that may have accumulated after S phase and necessary before cell division can continue.
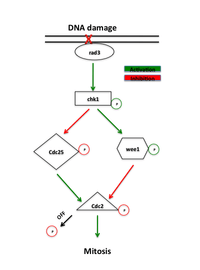
Proteins that function in the G2-M checkpoint were originally identified in yeast screens that looked for mutants which show enhanced sensitivity to radiation, termed "rad" mutants.[1] Inefficient repair of DNA damaged by ionizing radiation or chemical agents in these mutants revealed proteins essential in this pathway. Early signaling proteins in the checkpoint pathway are members of a family of phosphatidylinositol 3-kinases, rad3 in yeast and ATR in vertebrates, that are believed to localize to sites of DNA damage.[7] Rad3 phosphorylates rad26 which is required to initiate, but not maintain the checkpoint. Rad3 also phosphorylates a number of other proteins whose absence abolishes checkpoint DNA repair, including rad1, rad9, hus1 and rad17.[1] It has been hypothesized that rad9, hus1 and rad17 are similar to proteins involved in forming the clamp that increases the processivity of DNA polymerase during DNA replication.[8] In agreement with this idea, rad17 is similar to proteins involved in loading the clamp onto DNA. This supports a model where phosphorylation by rad3 causes recruitment of these proteins to sites of DNA damage where they mediate the activity of DNA polymerases involved in DNA repair.[1]
The main rad3 effector is the kinase Chk1, which is required for the G2-M arrest in response to DNA-damaging agents.[9] Chk1 is an effector protein kinase that maintains mitotic cyclin in an inactive state and is phosphorylated by rad3 between S phase and mitosis, implicating its specific role in G2 arrest.[10] Its upregulation through overexpression can induce arrest independent of DNA damage.[11] In addition, overexpression of Chk1 rescues the radiation sensitivity of rad mutants, presumably by allowing DNA repair to take place before entry into mitosis.[7]
The presence of DNA damage triggers the ATM (Ataxia telangiectasia mutated) or ATR (Ataxia Telangiectasia and Rad3 related) pathways which activate the Chk2 and Chk1 kinases, respectively. These kinases act upstream of Cdc25 and Wee1, the direct regulators of the CyclinB-Cdc2 complex. Chk1 and Chk2 phosphorylate Cdc25, inhibiting its phosphorylating activity and marking it for ubiquitinated degradation.[11][12] These pathways also stimulate the tumor suppressor p53. p53 regulates the function of the Cdk2 inhibitor p21 and the 14-3-3 proteins that phosphorylate (and thereby inactivate) and sequester Cdc25 in the cytoplasm, respectively.[13] Recent studies have also suggested that Cdk1 and 14-3-3 positively regulate Wee1 in a similar manner. The hyperphosphorylation of Wee1 by Cdk1 allows for the binding of 14-3-3, sequestering Wee1 to the nucleus and enhancing its ability to phosphorylate Cdc2.[14] The phosphorylation of both Wee1 and Cdc25 prevents Cdc2 activation.[12]
The ATM/ATR pathway also results in the negative regulation of Plk1 that contributes to the stability of Wee1. The stabilization of Wee1 and Myt1 ensures the cells arrest in G2 and allows for DNA repair.[13][15]
Multiple pathways are involved in the checkpoint response and thus, the targeting of Cdc25 is not the sole mechanism underlying cell cycle delay, as some models have proposed. The cooperativity between the positive regulation of Wee1 and the negative regulation of Cdc25 by Chk1 in response to unreplicated or damaged DNA results in a strong G2 arrest.[1][11][13][15] The increase in the amount of Wee1 and the decrease in the amount of Cdc25 contributes to the increase in the cyclin B concentration threshold in the hysteresis loop needed to drive the cell into mitosis.
Maintaining the Checkpoint
Rad3 is required for activation of Chk1 and initiation of G2 arrest, but different proteins are believed to maintain G2 arrest so that sufficient DNA repair can occur. One such protein is rad18 that is required for G2 arrest even when Chk1 is phosphorylated and active. Thus, rad18 is required for G2/M checkpoint maintenance while Chk1 is required for checkpoint initiation.[16] This is further supported by its additional function in DNA repair, specifically in the maintenance of chromosomal structures. Its necessity is demonstrated by the fact that in the absence of rad18, DNA is unable to be repaired even when G2 arrest is prolonged by other means.
The maintenance of such arrest in the G2 phase is further sustained by p53 and p21. In the absence of p53 or p21, it was demonstrated that radiated cells progressed into mitosis.[17] The absence of p21 or 14-3-3 cannot sufficiently inhibit the CyclinB-Cdc2 complex, thus exhibiting the regulatory control of p53 and p21 in the G2 checkpoint in response to DNA damage.[12] p53 mutations can result in a significant checkpoint deficit, which has important implications in the treatment of cancer.
Checkpoint Inactivation
Inactivation of both Wee1 and Cdc25 abolishes the G2-M DNA damage checkpoint. Absence of Wee1 or removal of the tyrosine-15 site removes negative regulation of Cdc2 activity and causes cells to enter mitosis without completing repair, which effectively abolishes the G2-M checkpoint.[18] Absence of Cdc25 arrests cells in G2, but still allows activation of the G2-M checkpoint, implicating that both the activation of Wee1 and deactivation of Cdc25 as important regulatory steps in the checkpoint.[11]
Inactivation of Chk1 is sufficient to surpass the checkpoint and promote entry into mitosis, regardless if DNA damage is repaired. Yet, little is still known about the exact mechanism regarding checkpoint termination with possible mechanisms including protein phosphatases reversing activating phosphorylations, targeted ubiquitin degradation of activating proteins, and checkpoint antagonists promoting mitosis through independent pathways.[10]
Cancer
Many cell cycle regulators like Cdks, cyclins, and p53 have been found to have abnormal expression in cancer. More specifically, they have been implicated in being involved in the G2/M transition by localizing to the centrosome, which thus leads to studies in manipulating such proteins in order to improve cancer’s sensitivity to radiation and chemotherapy.[13] Chk1 has important implications in drug targeting for cancer as its function acts in response to DNA damage. The cytotoxic effects of chemotherapy are currently being studied in the modulation of the G2/M transition, concerning both checkpoint abrogation or checkpoint arrest.[19] Many therapies focus on inactivating the checkpoint in order to force cells with excess DNA damage to proceed through mitosis and induce cell death.[12]
References
- Cuddihy, Andrew R.; O'Connell, Matthew J. (2003). "Cell-cycle responses to DNA damage in G2". International Review of Cytology. 222: 99–140. doi:10.1016/s0074-7696(02)22013-6. ISBN 9780123646262. ISSN 0074-7696. PMID 12503848.
- Morgan, David Owen, 1958- (2007). The cell cycle : principles of control. London: New Science Press. ISBN 978-0-19-920610-0. OCLC 70173205.CS1 maint: multiple names: authors list (link)
- Gould, K. L.; Nurse, P. (1989). "Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis". Nature. 342 (6245): 39–45. Bibcode:1989Natur.342...39G. doi:10.1038/342039a0. PMID 2682257.
- Seki, A.; Coppinger, J. A.; Jang, C.-Y.; Yates, J. R.; Fang, G. (20 June 2008). "Bora and the Kinase Aurora A Cooperatively Activate the Kinase Plk1 and Control Mitotic Entry". Science. 320 (5883): 1655–1658. Bibcode:2008Sci...320.1655S. doi:10.1126/science.1157425. PMC 2834883. PMID 18566290.
- Novak, B.; Tyson, J. J. (1993). "Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos". Journal of Cell Science. 106: 1153–1168. PMID 8126097.
- Sha, Wei; et al. (September 2002). "Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts". Proceedings of the National Academy of Sciences. 100 (3): 975–980. Bibcode:2003PNAS..100..975S. doi:10.1073/pnas.0235349100. PMC 298711. PMID 12509509.
- Al-Khodairy, F.; Carr, A. M. (1992). "DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe". The EMBO Journal. 11 (4): 1343–1350. doi:10.1002/j.1460-2075.1992.tb05179.x. PMC 556583. PMID 1563350.
- Thelen, M. P.; Venclovas, C.; Fidelis, K. (1999). "A sliding clamp model for the Rad1 family of cell cycle checkpoint proteins". Cell. 96 (6): 769–770. doi:10.1016/s0092-8674(00)80587-5. PMID 10102265.
- Walworth, N.; Davey, S.; Beach, D. (1993). "Fission yeast chkl protein kinase links the rad checkpoint pathway to cdc2". Nature. 363 (6427): 368–371. Bibcode:1993Natur.363..368W. doi:10.1038/363368a0. PMID 8497322.
- Calonge, T. M.; O'Connell, M. J. (2007). "Turning off the G2 DNA damage checkpoint". DNA Repair (Amst). 7 (2): 136–140. doi:10.1016/j.dnarep.2007.07.017. PMC 2233850. PMID 17851138.
- Raleigh, J. M.; O'Connell, M. J. (2000). "The G(2) DNA damage checkpoint targets both Wee1 and Cdc25". Journal of Cell Science. 113 (10): 1727–1736. PMID 10769204.
- Morgan, David (2007). The Cell Cycle Principles of Control. New Science Press. pp. 227–245.
- Wang, Y.; Ji, P.; Liu, J.; Broaddus, R. R.; Xue, F.; Zhang, W. (2009). "Centrosome-associated regulators of the G2/M checkpoint as targets for cancer therapy". Molecular Cancer. 8 (1): 8. doi:10.1186/1476-4598-8-8. PMC 2657106. PMID 19216791.
- Lee, J.; Kumagai, A.; Dunphy, W. G. (2001). "Positive regulation of Wee1 by Chk1 and 14-3-3 proteins". Molecular Biology of the Cell. 12 (3): 551–563. doi:10.1091/mbc.12.3.551. PMC 30963. PMID 11251070.
- Harper, J. W.; Elledge, S. J. (December 2007). "The DNA Damage Response: Ten Years After". Molecular Cell. 28 (5): 739–745. doi:10.1016/j.molcel.2007.11.015. PMID 18082599.
- Verkade, H. M.; Bugg, S. J.; Lindsay, H. D.; Carr, A. M.; O'Connell, M. J. (1999). "Rad18 is required for DNA repair and checkpoint responses in fission yeast". Molecular Biology of the Cell. 10 (9): 2905–2918. doi:10.1091/mbc.10.9.2905. PMC 25529. PMID 10473635.
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J. P.; Sedivy, J. M.; Kinzler, K. W.; Volgestein, B. (1998). "Requirement for p53 and p21 to Sustain G2 Arrest After DNA Damage". Science. 282 (5393): 1497–1501. doi:10.1126/science.282.5393.1497. PMID 9822382.
- Lundgren, K.; Walworth, N.; Booher, R.; Dembski, M.; Kirschner, M.; Beach, D. (1991). "Mik1 and wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2". Cell. 64 (6): 1111–1122. doi:10.1016/0092-8674(91)90266-2. PMID 1706223.
- DiPaola, R. S. (2002). "To Arrest or Not To G2-M Cell-Cycle Arrest". Clinical Cancer Research. 8 (11): 3311–3314.