Degos disease
Degos disease, also known as Köhlmeier-Degos disease or malignant atrophic papulosis, is an extremely rare condition caused by blockage of arteries and veins. Individuals with this condition will develop papules. Those diagnosed with this disease may also develop complications due to impairment of internal organs. The exact underlying mechanism is still unknown, and an effective treatment is still being developed.[1] There are fewer than 50 living patients presently known worldwide, and fewer than 200 reported in medical literature. However, many individuals may go undiagnosed due to rarity of the disease.[2][3] Most individuals develop symptoms between the ages of 20–50; however, cases outside of this age range have been reported as well.[1]
| Degos disease | |
|---|---|
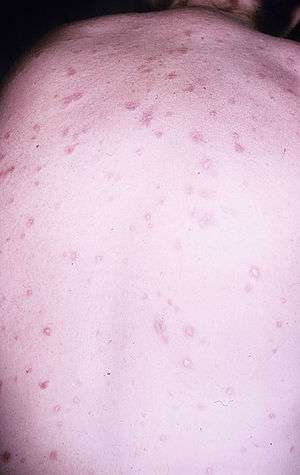 | |
| Skin lesions in a person with Degos disease | |
| Specialty | Cardiology, dermatology |
Symptoms
The characteristic symptom of Degos disease is the development of papules. Initially, individuals may have skin lesions or rashes, but they will proceed to develop distinct bumps, or papules.[4] Papules are circular in shape, have a porcelain-white center and red border. As papules age, the white centers will skin in and only the border will remain raised. Typically, papules range from 0.5 to 1 cm in width. Papules appear on the trunk and upper extremities and are not found on the individual's palms, soles, scalp, or face.[1]
Depending on whether an individual has the benign variant or malignant variant of the disease symptoms will vary. Both the benign and malignant forms have development of the characteristic papules. Individuals with the benign form will have the typical papules persisting anywhere from a few years to throughout their whole lives. In the benign form, no inner organs are affected. If an individual develops the malignant form, it means that not only are the papules present, but inner organs are involved. Most malignant cases involve problems of the gastrointestinal tract leading to small intestine lesions, abdominal pain, diarrhea, and bowel perforation. If the central nervous system is involved, symptoms can include headaches, dizziness, seizures, paralysis of cranial nerves, weakness, stroke, damage to small areas of the brain due to artery blockage (cerebral infarcts, and cerebral hemorrhage). Additional organs commonly impacted include the heart, lungs, and kidneys. Symptoms that may develop from damage to these organs include double vision (diplopia), clouding of lenses of eyes, swelling of the optic disc (papilledema), partial loss of vision, shortness of breath, chest pain, epilepsy, and thickening of pericardium.[3][4]
Someone with the benign form may suddenly develop symptoms of the malignant form.[1] Symptoms can last anywhere from a few weeks to several years. Onset of symptoms typically begins to manifest between the ages of 20–50.[5] A few cases of this condition in newborns have also been described.[6]
Causes
The papules characteristic for this disease develop due to infarctions, or blockages in small-medium arteries and veins. The underlying cause is unknown for this disease.[1] Though not confirmed, some cases have shown signs of inheritance between first-degree relatives. It has been suggested that the disease has a familial inheritance pattern; it is thought to be an autosomal dominant disorder. In most cases of familial inheritance, the benign variant of the disease has been present.[4][7][8]
Due to the lack of knowledge of the pathomechanism for this condition prevention strategies are not known. However, in order to prevent worsening of symptoms, consistent evaluations should be conducted by a physician.[1]
Mechanism
Although this disease has been known for around 70 years, the pathomechanism underlying it is still unknown.[1] Several hypotheses have been developed regarding the underlying mechanism for Degos disease. One theory suggests that inflammation of blood vessels may trigger development of the condition. Another theory has to do with Degos disease as a coagulopathy. Development of a thrombus and resulting reduction of blood flow is common in this condition. A reduction in blood flow throughout the body can lead to damaged endothelial cells and may perhaps lead to the formation of the characteristic papules. Another hypothesis suggests that abnormal swelling and proliferation of the vascular endothelium can lead to intestinal and central nervous system thrombosis, and ultimately lead to development of symptoms associated with Degos disease. Overall, individuals with Degos disease have abnormal blockages in their arteries and veins; however, the cause of these blockages is unknown.[1]
Diagnosis
Clinical evaluation and identification of characteristics papules may allow a dermatologist to diagnose Degos disease.[4] The papules have a white center and are bordered with a red ring. After lesions begin to appear, the diagnosis for Degos disease can be supported by histological findings. Most cases will show a wedge-shaped connective tissue necrosis in the deep corium. This shape is due to the blockage/occlusion of small arteries.[1]
Individuals may be diagnosed with the benign form if only the papules are present. However, an individual may be diagnosed with the malignant form if involvement of other organs like the lungs, intestine and/or central nervous system occurs. The malignant, or systematic form of this condition may suddenly develop even after having papules present for several years. In order to quickly diagnose this shift to the malignant variant of the disease, it is important for individuals to have consistent follow-up evaluations. In these evaluations, depending on which organs are suspected to be involved, the following procedures and tests may be conducted: skin inspection, brain magnetic resonance tomography, colonoscopy, chest X-ray, and/or abdominal ultrasound.[1]
Treatment
Due to the lack of knowledge around the underlying mechanism of malignant atrophic papulosis, an effective treatment method has not been developed.[1][1] Treatment for this condition is symptomatic.[4] However, several treatment methods have been tested and are still being developed as more information regarding the condition is found. Fibrinolytic and immunosuppressive therapeutic regimens were tested and found to be mostly unsuccessful as treatment methods.[1][8]
After treating conditions comorbid with Degos disease, physicians have recently found improvement in symptoms with the use of eculizumab and treprostinil.[9][10] Discovered by dermatopathologist, Cynthia Magro, response to eculizumab is often immediate and dramatic, but has been of limited duration and is expensive, needing to be infused every 14 days.[9] Treprostinil use has been reported to result in clearing of gastrointestinal and central nervous system findings as well as clearing of cutaneous lesions, but reports are limited. Treprostinil may be more effective than other vasodilators because it may also increase the population of circulating endothelial cells, allowing angiogenesis.[1]
Recent research
A 46-year-old male patient was diagnosed with the malignant, systemic form of the disease and was severely ill. The diagnosing dermatopathologist, Cynthia Magro MD, identified the presence of C5b-9 complexes in the involved vessels of the skin biopsy. For treatment of the thrombotic microangiopathy in this patient, she suggested the use of eculizumab, a humanized monoclonal antibody drug developed by Alexion Pharmaceuticals and approved by the Food and Drug Administration for treatment of Paroxysmal nocturnal hemoglobinuria. The patient experienced a dramatic improvement in his condition.[9] Lee Shapiro MD and Aixa Toledo-Garcia MD at Albany Medical College learned of the success with the adult patient, and became the first physicians to successfully treat a pediatric Degos patient with eculizumab.[10]
Dr. Shapiro later observed the resolution of Degos skin lesions in an adult patient with an overlap syndrome involving systemic lupus, systemic sclerosis, and Degos disease who was treated with treprostinil for her pulmonary hypertension. His pediatric Degos patient was developing significant complications despite treatment with eculizumab, so Dr. Shapiro's group became the first to treat a Degos patient with treprostinil.[10] To this point, all known long-term survivors of systemic Degos disease are being treated with a combination of eculizumab and treprostinil.[9][10]
History
In 1941, this disease was first described by Kohlmeier. However, it was not until 1942 that the disease was recognized as a new clinical entity by Robert Degos. Initially the condition was referred to as Degos Disease or Kohlmeier-Degos disease. However, Degos himself subsequently suggested the name "papulose atrophiante maligne," translated as malignant atrophic papulosis.[11]
References
Notes
- Theodoridis, Athanasios; Makrantonaki, Evgenia; Zouboulis, Christos C. (2013-01-14). "Malignant atrophic papulosis (Köhlmeier-Degos disease) - A review". Orphanet Journal of Rare Diseases. 8: 10. doi:10.1186/1750-1172-8-10. ISSN 1750-1172. PMC 3566938. PMID 23316694.
- "Degos Disease: Background, Pathophysiology, Epidemiology". 2017-02-07. Cite journal requires
|journal=(help) - De Breucker S, Vandergheynst F, Decaux G (2008). "Inefficacy of intravenous immunoglobulins and infliximab in Degos' disease". Acta Clin Belg. 63 (2): 99–102. doi:10.1179/acb.2008.63.2.007. PMID 18575050.
- "Degos Disease - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). Retrieved 2017-12-13.
- Snow, J. L.; Muller, S. A. (June 1995). "Degos syndrome: malignant atrophic papulosis". Seminars in Dermatology. 14 (2): 99–105. doi:10.1016/S1085-5629(05)80004-5. ISSN 0278-145X. PMID 7640203.
- Torrelo, A.; Sevilla, J.; Mediero, I.g.; Candelas, D.; Zambrano, A. (2002-05-01). "Malignant atrophic papulosis in an infant". British Journal of Dermatology. 146 (5): 916–918. doi:10.1046/j.1365-2133.2002.04677.x. ISSN 1365-2133.
- Katz, Sara K.; Mudd, Leslie J.; Roenigk, Henry H. (1997). "Malignant atrophic papulosis (Degos' disease) involving three generations of a family". Journal of the American Academy of Dermatology. 37 (3): 480–484. doi:10.1016/s0190-9622(97)70151-8.
- Powell; Bordea; Wojnarowska; Farrell; Morris (1999-09-01). "Benign familial Degos disease worsening during immunosuppression". British Journal of Dermatology. 141 (3): 524–527. doi:10.1046/j.1365-2133.1999.03050.x. ISSN 1365-2133.
- Magro, CM; Poe, JC; Kim, C; Shapiro, L; Nuovo, G; Crow, MK; Crow, YJ (2011). "Degos disease: A C5b-9/interferon-α-mediated endotheliopathy syndrome". Am J Clin Pathol. 135 (4): 599–610. doi:10.1309/ajcp66qimfarlzki. PMID 21411783.
- Shapiro, LS; Toledo-Garcia, AE; Farrell, JS (April 4, 2013). "Effective treatment of malignant atrophic papulosis (Köhlmeier-Degos disease) with treprostinil--early experience". Orphanet J Rare Dis. 8: 52. doi:10.1186/1750-1172-8-52. PMC 3636001. PMID 23557362.
- Degos R.; Delort J.; Tricot R. (1942). "Dermatite papulosquameuse atrophiante". Bulletin de la Société Française de Dermatologie et de Syphiligraphie et de Ses Filiales. 49: 148–150.
Further reading
- Scheinfeld N (September 2007). "Malignant atrophic papulosis". Clin. Exp. Dermatol. 32 (5): 483–7. doi:10.1111/j.1365-2230.2007.02497.x. PMID 17692056.
- Fazio S (June 13, 2014). "Rash, Myalgia, and Weakness". Now@NEJM.
External links
| Classification | |
|---|---|
| External resources |